Capítol 4 Enllaç Químic i Forces Intermoleculars
(darrera actualització: 13 de maig de 2026)
Índex
4.1 Introducció
Sovint, les observacions macroscòpiques fetes amb els sentits estan arrelades en els tipus i les intensitats de les atraccions/repulsions entre espècies químiques a escala molecular. Per exemple, els teus ulls et diuen que encerar un cotxe fa que l’aigua formi grans gotes anomenades perles, però com ho fa, i com protegeix la cera el cos del cotxe de l’oxidació? Per què els sabons fan escuma i per què hi ha sabons i detergents especialitzats per a l’automòbil? Per què hi ha diferents ”pesos”per a l’oli de motor i per què el teu cotxe necessita un en particular? Totes aquestes preguntes estan relacionades amb les forces que es desenvolupen entre molècules en vapors, líquids i superfícies sòlides, a diferència de les interaccions d’enllaç, que ocorren dins d’una molècula[1]:
-
• Per a un àtom individual i per a les reaccions químiques, les forces d’enllaç dins de les molècules són importants. És l’energia emmagatzemada en els enllaços químics la que fa que els hidrocarburs com l’octà siguin excel·lents combustibles per al motor.
-
• Per entendre grans col·leccions de molècules que podem veure, agafar i tocar, les forces intermoleculars esdevenen més importants. Per exemple, les forces intermoleculars molt fortes entre les molècules d’H2O expliquen el punt d’ebullició anòmalament alt d’aquesta petita molècula, a causa dels ponts d’hidrogen que forma i del seu moment dipolar. Aquestes forces són més febles que els enllaços covalents o iònics, però són essencials per entendre la conducta de les substàncies en estat líquid i sòlid.
Moment dipolar
El moment dipolar és una mesura de la separació de càrrega en una molècula. Un dipol és una disposició de càrregues  i
i  separades per una distància (vegeu la Figura 4.1). Un dipol es defineix com una càrrega en un extrem i exactament la mateixa càrrega però oposada a l’altre extrem.
separades per una distància (vegeu la Figura 4.1). Un dipol es defineix com una càrrega en un extrem i exactament la mateixa càrrega però oposada a l’altre extrem.
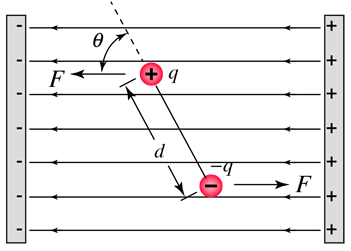
Avaluem l’energia d’un dipol en un camp elèctric. La Figura 4.1 mostra un dipol amb el seu centre fixat a l’espai. El dipol està subjecte a una força restauradora d’un camp elèctric  . Computem el treball
. Computem el treball  que el camp efectua en rotar el dipol des de
que el camp efectua en rotar el dipol des de  (paral·lel al camp) fins a un angle
(paral·lel al camp) fins a un angle  :
:
![\[ w = \int \vec {F} \cdot d\vec {l} = -2Eqa \int _{0}^{\theta } \sin \theta ' d\theta ' = Eqa (\cos \theta - 1). \]](web_GEAForcesInt-images/image-8.svg)
Aquesta equació és útil per modelitzar molècules polars orientant-se en camps elèctrics.
4.2 L’enllaç químic
Què entenem per molècula? Què és un enllaç? De quins tipus n’existeixen?
4.2.1 Tipus d’enllaços
És útil usar uns models conceptuals que expliquin l’estructura molecular. Són models extrems que no sempre es segueixen de manera exacta per les substàncies químiques. Sovint tenim barreges d’aquests models en un sistema real, però serveixen per conceptualitzar la manera en què la matèria s’organitza a nivell molecular i atòmic.
Els enllaços químics són les forces que mantenen els àtoms units en una molècula. Hi ha tres tipus d’enllaços químics: enllaços covalents, enllaços iònics i enllaços metàl·lics. La taula 4.1 mostra les característiques de cada tipus d’enllaç.

La majoria dels enllaços químics tenen propietats intermèdies entre el covalent i l’iònic però estan força aprop d’algun dels dos models.
4.2.2 Enllaç covalent
Els enllaços covalents es formen quan dos àtoms comparteixen electrons. Aquest tipus d’enllaç és molt comú en molècules orgàniques i inorgàniques. Els enllaços covalents poden ser simples, dobles o triples, depenent del nombre d’electrons compartits. Els enllaços covalents poden ser polars o no polars. En un enllaç covalent polar, els electrons es comparteixen de manera desigual entre els àtoms. Això es deu a la diferència d’electronegativitat entre els àtoms. En un enllaç covalent no polar, els electrons es comparteixen de manera igual entre els àtoms. Els enllaços covalents no polars són més forts que els enllaços covalents polars. Els enllaços covalents polars són més forts que els enllaços iònics. Els enllaços covalents són els més forts dels tres tipus d’enllaços químics.
Els enllaços vibren constantment per la  no nul·la de les molècules. No obstant això, podem mesurar distàncies d’enllaç promig que es pot mesurar a partir d’estudis de raigs X o l’espectroscopia molecular (Taula 4.2).
no nul·la de les molècules. No obstant això, podem mesurar distàncies d’enllaç promig que es pot mesurar a partir d’estudis de raigs X o l’espectroscopia molecular (Taula 4.2).
| Enllaç | BD / Å | Enllaç | BD / Å |
 |
1.42 |  |
0.92 |
 |
1.99 |  |
1.27 |
 |
2.28 |  |
1.41 |
 |
2.67 |  |
1.61 |
 |
1.63 |  |
0.74 |
 |
2.14 |  |
1.094 |
 |
1.76 |  |
1.207 |
 |
2.32 |  |
1.151 |
 |
1.128 |
Els valors de la taula són molt constants a totes les molècules que contenen aquests enllaços, fins i tot més que no pas el que succeix amb les energies d’enllaç promig de les Taules ?? i ??. Quan això no es
compleix és perquè hi ha enllaços diferents (dobles, triples...). Per exemple, entre l’età ( ), l’etilè (
), l’etilè ( ) i l’acetilè (
) i l’acetilè ( ) les energies d’enllaç entre els àtoms de C varien de 83 a 146 i 200 kcal mol
) les energies d’enllaç entre els àtoms de C varien de 83 a 146 i 200 kcal mol , i les distàncies de 1.54 a 1.34 i 1.20, respectivament.
, i les distàncies de 1.54 a 1.34 i 1.20, respectivament.
També se’n té constància en les geometries de les molècules, com s’aprecia a la Figura 4.2.
4.2.3 Enllaç iònic
Els enllaços iònics es formen quan un àtom cedeix electrons a un altre àtom. Aquest tipus d’enllaç és comú en compostos iònics com el clorur de sodi (NaCl). En un enllaç iònic, un àtom es converteix en un ió positiu (catió) i l’altre àtom es converteix en un ió negatiu (anió).
4.2.3.1 Formulació de Lewis d’un enllaç iònic
En termes de Lewis, la formació d’un enllaç iònic sorgeix de la transferència d’electrons d’un àtom a un altre. Quan es produeix aquesta transferència, tots els electrons de valència de l’element més electropositiu (d’un dels tres primers grups de l’esquerra de la taula periòdica) són eliminats per exposar el nucli de l’àtom. Els electrons alliberats són acceptats als orbitals buits de la capa de valència de l’àtom més electronegatiu (normalment dels grups immediatament a l’esquerra dels gasos nobles), omplint així la capa de valència.
Què són les estructures de Lewis?
Les estructures de Lewis són representacions esquemàtiques que mostren quins electrons de valència participen en els enllaços i quins queden com a parells no enllaçants. Serveixen per visualitzar de manera ràpida la connectivitat d’una espècie química i per fer una primera estimació de càrregues formals, geometries i reactivitat.
En aquestes representacions:
-
• un enllaç covalent es dibuixa com un parell d’electrons compartit, per exemple
 o, més habitualment,
o, més habitualment,  ;
;
-
• els parells solitaris es dibuixen com a punts al voltant de l’àtom;
-
• en ions o espècies poliatòmiques, es poden indicar les càrregues formals entre claudàtors, per exemple
![\ch{[NH4+]}](web_GEAForcesInt-images/379DE7A04CFA5AE1EE5825BE1AAC8156.svg) o
o ![\ch{[OH-]}](web_GEAForcesInt-images/DD999F213FE3BD64C85BA57F926D5222.svg) .
.
Per construir una estructura de Lewis acostumem a seguir quatre passos:
-
1. Comptar els electrons de valència totals de l’espècie.
-
2. Triar un esquelet raonable d’enllaços entre els àtoms.
-
3. Repartir els electrons restants per completar duets o octets quan sigui possible.
-
4. Revisar si les càrregues formals són plausibles i si hi ha formes ressonants rellevants.
Què és la capa de valència?
La capa de valència és la capa electrònica més externa ocupada d’un àtom. Els electrons que hi ha en aquesta capa són els que participen amb més facilitat en els enllaços químics, perquè són els que es troben més allunyats del nucli i, per tant, estan menys fortament retinguts. Per això, quan representem estructures de Lewis, només dibuixem els electrons de valència.
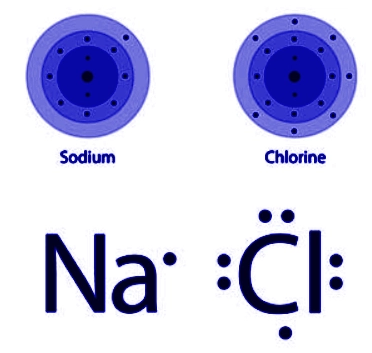
Així, la formació del compost iònic clorur de sodi es pot representar pel següent procés:



La Figura 4.3 representa aquesta transferència d’electrons amb la notació de Lewis.
La formació de l’òxid d’alumini (alúmina) implica seleccionar prou àtoms d’alumini i oxigen per assegurar que tots els electrons alliberats pels àtoms d’alumini (tres de cadascun) siguin acomodats pels àtoms d’oxigen (cadascun dels quals pot acceptar dos electrons):

El nombre d’àtoms requerits per equilibrar els electrons donats i acceptats s’indica en la fórmula química  per a l’òxid d’alumini.
per a l’òxid d’alumini.
4.2.3.2 L’Energia en la Formació d’Enllaços Iònics
El fet que la transferència d’electrons representada en aquests diagrames condueixi a una disminució de l’energia es pot verificar avaluant les energies associades al procés. L’energia total de formació d’un  ió i un
ió i un  ió és la suma de tres termes[3]:
ió és la suma de tres termes[3]:
-
1. L’energia d’ionització necessària per ionitzar un àtom de sodi.
-
2. L’energia alliberada quan un electró del sodi s’adhereix a un àtom de clor.
-
3. L’energia d’interacció entre el catió i l’anió, anomenada energia de xarxa.
Aquesta última contribució és fonamental, ja que compensa el cost energètic de la ionització del sodi i l’atracció entre les càrregues oposades fa que el parell iònic / , i encara més la xarxa  , siguin més estables que els àtoms neutres.
, siguin més estables que els àtoms neutres.
Tot i que, com veurem més endavant, l’estructura electrònica d’un àtom és complexa, podem pensar que des de la distància la distribució dels electrons segueix una forma propera a esfèrica. Per tant, i seguint la llei de Coulomb, aquestes esferes es comporten com si la seva càrrega estigués concentrada al seu centre, de manera que podem considerar els ions com a càrregues puntuals.
En el cas del clorur de sodi ( ), l’espectroscopia de raigs X mostra que l’estructura del compost és regular amb esferes que contenen 10 i 18 e
), l’espectroscopia de raigs X mostra que l’estructura del compost és regular amb esferes que contenen 10 i 18 e cadascuna, corresponents als ions i , respectivament. Això demostra que els ions existeixen i que, per tant, les forces que uneixen aquests ions han de ser, per força, elèctriques.
cadascuna, corresponents als ions i , respectivament. Això demostra que els ions existeixen i que, per tant, les forces que uneixen aquests ions han de ser, per força, elèctriques.
4.2.3.3 El Cicle de Born-Haber
L’anàlisi de la formació d’un compost iònic a partir dels seus elements es tracta comunament en termes d’un cicle de Born-Haber, que desglossa el procés en una sèrie de passos amb energia coneguda. Per exemple, per a la formació d’un mol de a partir de  i
i  , els passos rellevants són:
, els passos rellevants són:
-
1. L’atomització del sodi:
-
2. La semidissociació del clor:
-
3. Ionització del sodi:
-
4. Afinitat electrònica del clor:
-
5. Formació de la xarxa cristal·lina:





L’energia de xarxa és tan gran que més energia és alliberada en aquest pas que la necessària per als passos anteriors combinats, fent que el sòlid tingui menor energia que el sodi metàl·lic i el clor gasós.

Tenint en compte altres dades experimentals, podem veure que la formació d’un mol de molècules de implica les següents relacions energètiques:
![\[ \begin {array}{cc} \begin {array}{c} \ch {Na(g) -> Na+ (g) + e^-}\\ \ch {Cl(g) + e^- -> Cl^- (g)}\\ \hline \ch {Na(g) + Cl(g) -> Na+ (g) + Cl^- (g)}\\ \\ \ch {Na+ (g) + Cl^- (g) -> NaCl(g)}\\ \hline \ch {Na(g) +
Cl(g) -> NaCl(g)} \end {array} \quad \begin {array}{c} \Delta E=PI(\ch {Na}) = \qty {118.4}{\kcal \per \mole } \\ \Delta E=-AE(\ch {Cl}) = \qty {-83.4}{\kcal \per \mole }\\ \hline \Delta E= \qty {35}{\kcal \per \mole } \\ \\ \Delta E =
\qty {-139.3}{\kcal \per \mole }\\ \hline \Delta E = \qty {-104.3}{\kcal \per \mole } \end {array} \end {array} \]](web_GEAForcesInt-images/image-65.svg)
L’esquema mostra que la formació dels ions gasosos, per si sola, no és favorable. El procés esdevé globalment favorable quan incorporem l’estabilització electrostàtica associada a la formació del parell iònic i, sobretot, de la xarxa cristal·lina.
No obstant això, ens interessa entendre com es formen els cristalls de . Aquests cristalls tenen pressions de vapor extremadament baixes i, per tant, difícilment trobarem com a espècie gasosa. Per calcular quanta energia es desprèn en formar el sòlid hem de tenir en compte l’entalpia de malla  , que en aquest context correspon a l’entalpia molar estàndard del procés
, que en aquest context correspon a l’entalpia molar estàndard del procés  a 1 atm i 0 °C. A 0 K,
a 1 atm i 0 °C. A 0 K,  , l’energia de malla, que només depèn de les interaccions coulombianes entre ions. A temperatures habituals, la diferència entre totes dues magnituds és relativament petita.
, l’energia de malla, que només depèn de les interaccions coulombianes entre ions. A temperatures habituals, la diferència entre totes dues magnituds és relativament petita.
Fem un càlcul d’aquesta energia potencial. Imaginem una disposició lineal d’ions positius i negatius amb càrregues  i
i  , respectivament, separats per una distància
, respectivament, separats per una distància  . L’energia potencial del primer ió seria:
. L’energia potencial del primer ió seria:

Quantitat que haurem de multiplicar per 2 per tal de considerar els dos costats de l’ió, així com per  per tal d’obtenir el valor molar. Finalment, podríem generalitzar el resultat per a qualsevol xarxa d’ions de càrregues de diferent signe
per tal d’obtenir el valor molar. Finalment, podríem generalitzar el resultat per a qualsevol xarxa d’ions de càrregues de diferent signe  i
i  , tot obtenint el resultat:
, tot obtenint el resultat:
![\[ E_p=-A\frac {|z_A z_B|N_{\mathrm {A}} e^2}{4\pi \varepsilon _0 d} \]](web_GEAForcesInt-images/image-78.svg)
on  és la constant de Madelung, que depèn de l’estructura tridimensional del cristall (per al ,
és la constant de Madelung, que depèn de l’estructura tridimensional del cristall (per al ,  ).
).
No obstant això, aquesta no és l’única contribució a l’energia de malla, ja que cal incorporar el solapament que es produeix entre els orbitals dels dos ions quan s’apropen. Aquesta contribució és proporcional al factor  , on
, on  pren un valor aproximat de 34,5 pm.
pren un valor aproximat de 34,5 pm.
Si sumem les dues contribucions i trobant-ne el mínim, obtenim l’equació de Born-Mayer:

Exercici 1. Equació de Born-Mayer
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Dedueix l’equació de Born-Mayer a partir de considerar, de forma simplificada, que l’energia d’atracció Coulòmbica es pot expressar com  i que la repulsió entre ions es pot expressar com
i que la repulsió entre ions es pot expressar com  .
.
Exercici 2. Energia de malla
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
L’òxid de magnesi,  , té la mateixa estructura que el . Sabent que
, té la mateixa estructura que el . Sabent que  i que
i que  , calcula l’energia de malla d’aquest compost iònic.
, calcula l’energia de malla d’aquest compost iònic.
4.2.3.4 Propietats dels sòlids iònics
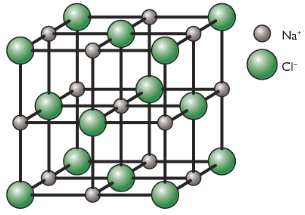
Els sòlids iònics són compostos formats per ions que es mantenen units per forces electrostàtiques. Aquestes forces són molt fortes, de manera que els sòlids iònics tenen punts de fusió i ebullició molt alts. Els sòlids iònics són generalment durs i fràgils, i no condueixen l’electricitat en estat sòlid, però sí quan es fonen o es dissolen en aigua.
Un sòlid iònic és una xarxa tridimensional de cations i anions atrets per forces electrostàtiques. Aquest és el cas, per exemple, del NaCl, CaF o CsO, en què la diferència d’electronegativitats entre els elements és molt gran. En canvi, si aquesta diferència és menor (p. ex. NB), es diu que l’enllaç iònic presenta un cert caràcter covalent.
o CsO, en què la diferència d’electronegativitats entre els elements és molt gran. En canvi, si aquesta diferència és menor (p. ex. NB), es diu que l’enllaç iònic presenta un cert caràcter covalent.
-
• En un sòlid iònic ideal, els electrons es troben totalment localitzats en els seus ions respectius, de manera que no hi ha una localització electrònica com ens passa en els sòlids covalents. És precisament aquest el motiu pel qual els sòlids iònics no condueixen el corrent (només el condueixen quan estan fosos).
-
• Els sòlids iònics són generalment menys densos, menys durs i més fràgils i trencadissos que els metalls. Per això no són mecanitzables com els metalls.
-
• Els sòlids iònics són generalment solubles en aigua (p. ex. halita o NaCl, CaCl
), però hi ha excepcions (la fluorita o CaF és molt insoluble en aigua).
Els sòlids iònics són sòlids cristal·lins que compleixen:
-
1. Cada catió té al seu voltant el màxim nombre possible d’anions i cada anió el màxim nombre possible de cations (màxim nombre de coordinació (N.C.) possibles per a cations i anions).
-
2. La separació entre ions de la mateixa càrrega és la màxima possible i entre ions de càrrega oposada la mínima possible (generalment hi ha contactes directes anió-catió).
-
3. La proporció entre el nombre d’anions i cations ha de correspondre a l’estequiometria del sòlid.
La major part d’estructures dels sòlids iònics es poden considerar derivades d’un empaquetament més o menys compacte d’anions amb els cations ocupant tots o una part dels forats que deixen els anions, ja que generalment els cations són més petits que els anions. Segons les regles de Pauling:
-
1. Al voltant de cada catió, es forma un poliedre de coordinació d’anions. La distància catió-anió en aquest poliedre es determina per la suma dels radis del catió i l’anió, i el nombre de coordinació es determina pel ratio entre els radis del catió i l’anió. El nombre de coordinació augmenta a mesura que el ratio de radis augmenta, amb diferents geometries corresponents a diferents nombres de coordinació.
-
2. La compartició d’arestes i, especialment, de cares entre dos poliedres d’anions en una estructura cristal·lina disminueix la seva estabilitat.
-
3. En una estructura cristal·lina que conté diversos cations, aquells amb alta valència i un nombre de coordinació petit tendeixen a no compartir elements polièdrics.
-
4. El principi de la parsimònia. El nombre de diferents tipus de constituents en un cristall tendeix a ser petit.
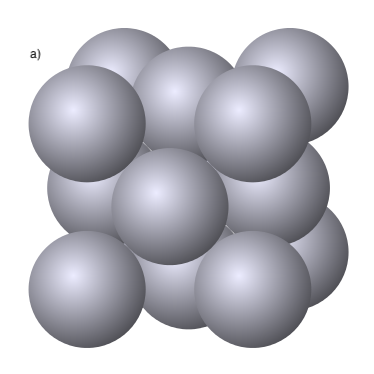
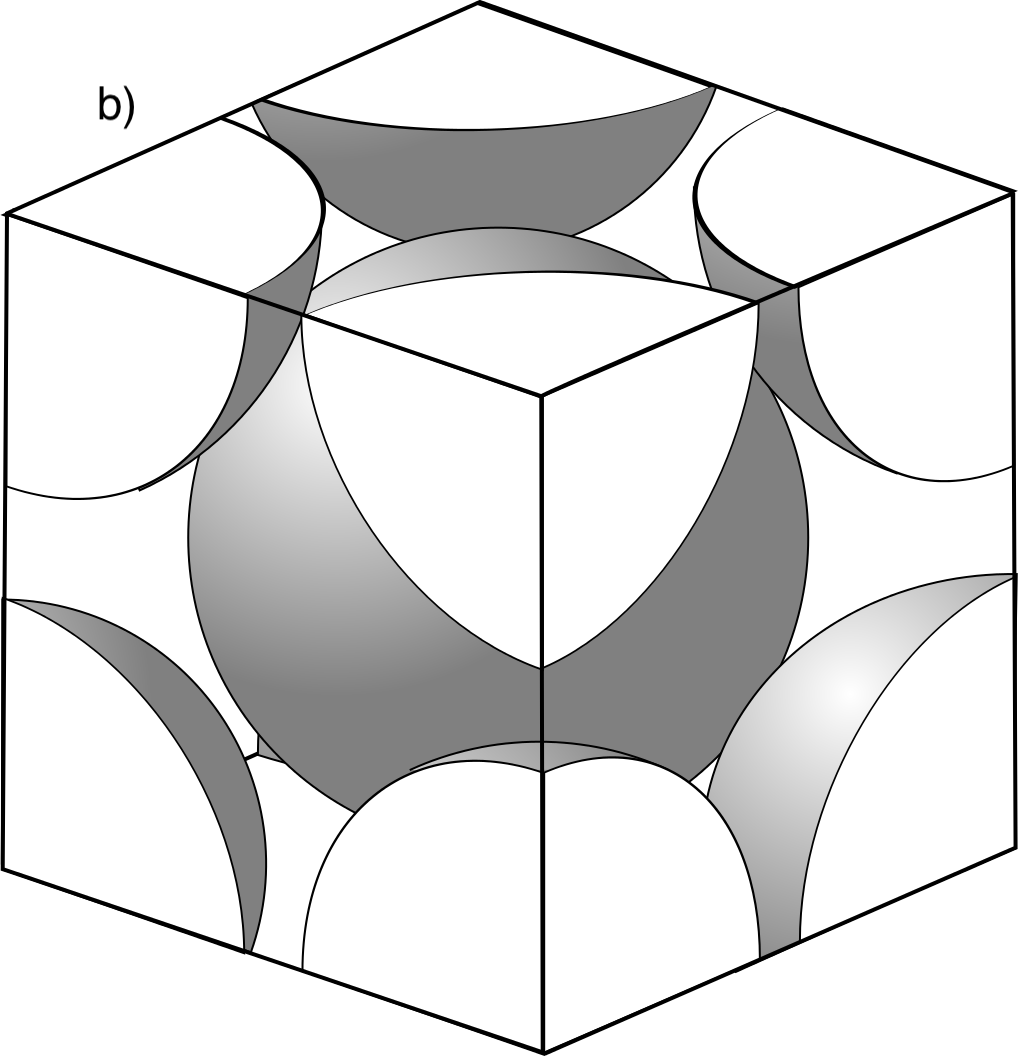
La Figura 4.4 mostra tres cel·les unitat habituals que apareixen sovint en metalls i sòlids iònics.
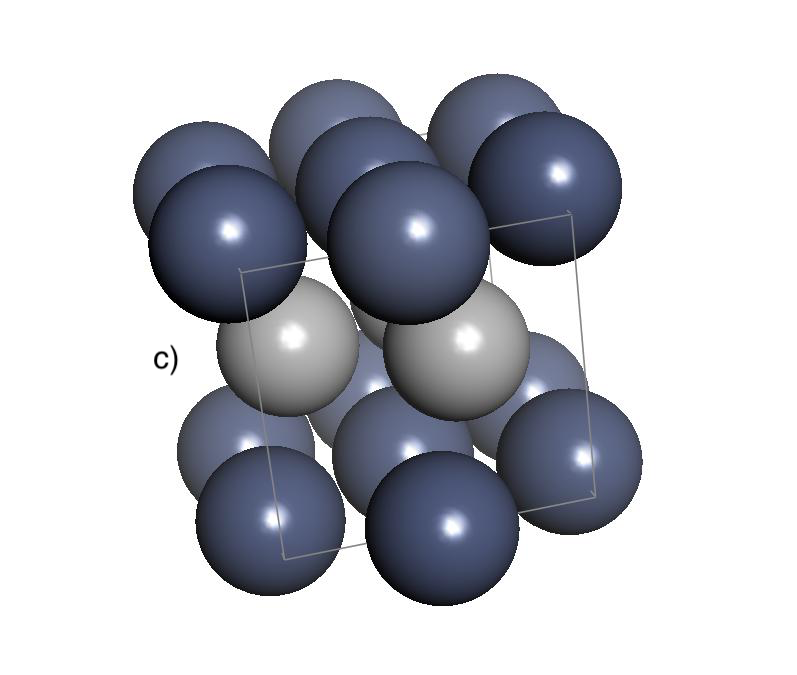










|
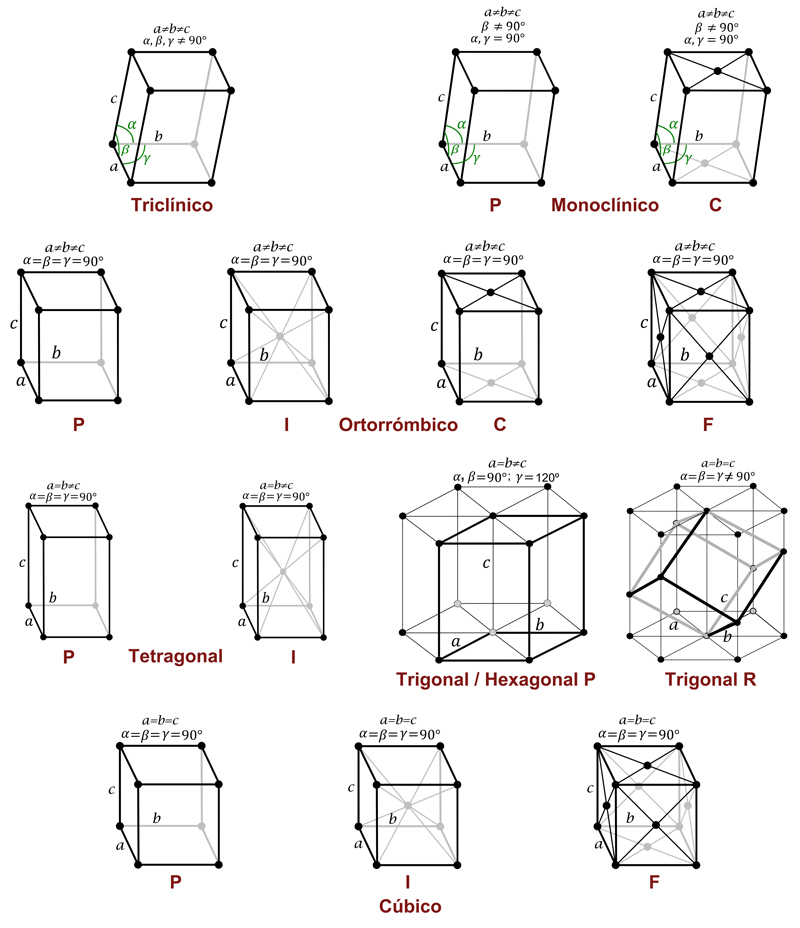
Taula 4.4: Sistemes cristal·lins i retícules de Bravais. P: centrada en les cantonades; I: centrada en el cos; F: centrada en la cara; C: amb punt central (adaptat de [16]). |
||
| Sistema | Cel·la unitat | Retícula de Bravais |
|
Taula 4.4: (Continuació) |
||
| Sistema | Cel·la unitat | Retícula de Bravais |
| Cúbic |  |
P, I (Fig. 4.4b), F (Fig. 4.4a) |
 |
||
| Tetragonal |  |
P, I |
|
||
| Ortoròmbic |  |
P, I, C, F |
|
||
| Romboèdric | |
R(P) |
 |
||
| Hexagonal | |
P (Fig. 4.4c) |
 |
||
 |
||
| Monoclínic |  |
P, C |
 |
||
| Triclínic | |
P |
 |
||
La Figura 4.5 recull les retícules de Bravais associades als set sistemes cristal·lins.
4.2.3.5 Defectes
Fins ara hem suposat que els cristalls són perfectes. En realitat, tots els cristalls presenten defectes, els quals afecten lleugerament la densitat, la capacitat calorífica i l’entropia dels cristalls, però alteren profundament la resistència mecànica, la conductivitat elèctrica, la velocitat de difusió i l’activitat catalítica. Les imperfeccions en els sòlids es classifiquen en defectes puntuals, lineals o de plans[6].
-
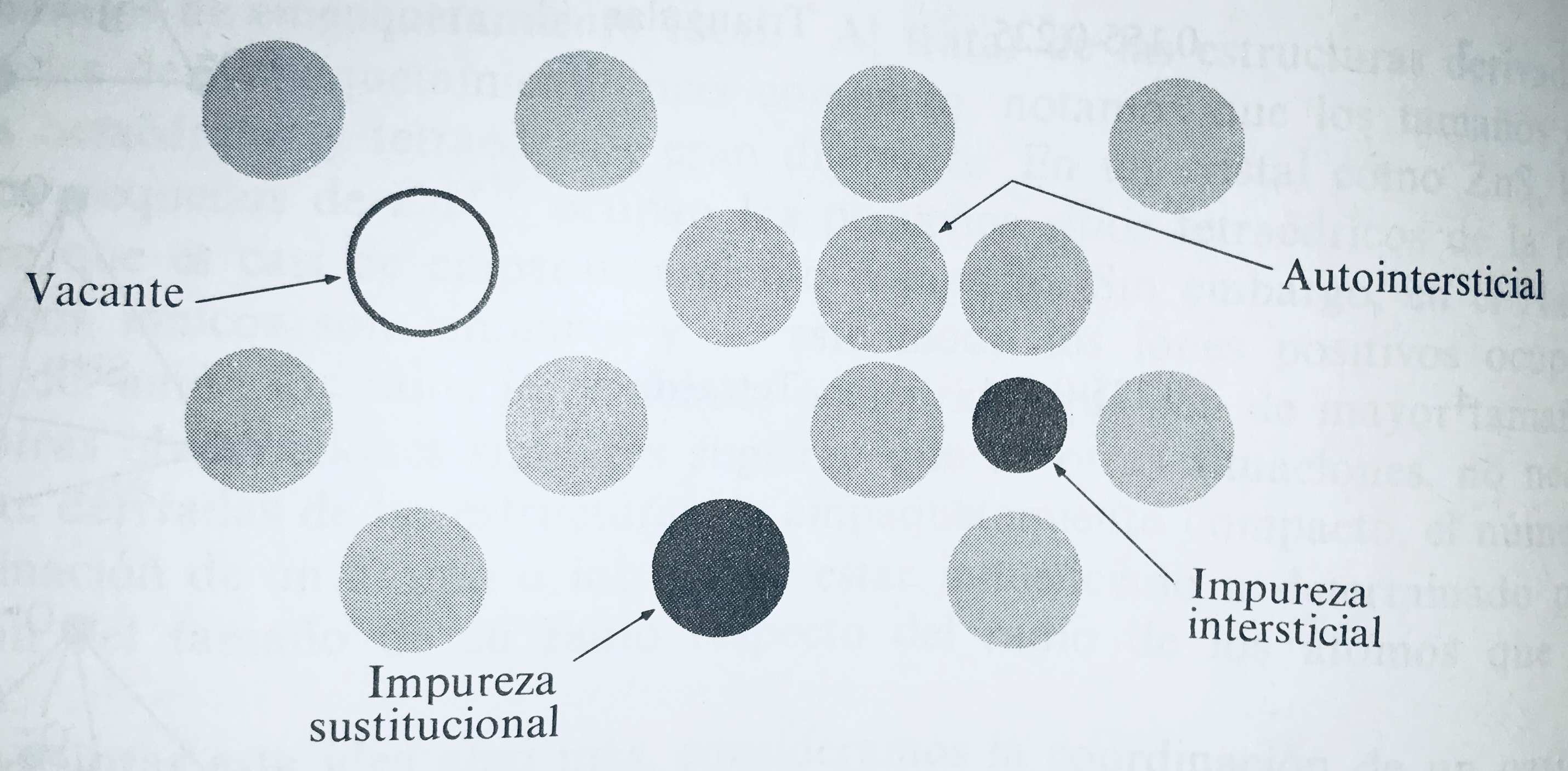
• Defectes de punt: Impliquen una sola posició (veure Figura 4.6). Un forat és l’absència d’un àtom, ió o molècula en un lloc que hauria d’estar ocupat en un cristall perfecte. Una impuresa substitucional és una impuresa (àtom, molècula o ió) situada en un lloc ocupat per una altra espècie en un cristall perfecte; una impuresa intersticial està situada en un lloc (buit) que estaria desocupat en un cristall perfecte. Un intersticial propi és una no impuresa (àtom, molècula o ió) situada en un buit. Quan augmenta la temperatura del cristall, el nombre d’àtoms, molècules o ions que tenen suficient energia vibracional per sortir dels seus llocs en el cristall perfecte augmenta, incrementant així el nombre de forats i d’intersticials propis.
Identifiquem els defectes de Schottky com aquells on apareixen vacants catió-anió en parelles. En un defecte de Frenkel, en canvi, hi ha un desplaçament d’un catió cap a una posició intersticial. En els dos casos es manté la neutralitat de l’estructura (a la fluorita, per exemple, els intersticis són grans, i per tant és fàcil trobar defectes de Frenkel). Si el defecte és l’absència d’un anió podem tenir un defecte de tipus centre F.
Els llocs o centres catalítics de les superfícies dels òxids metàl·lics són normalment deguts a forats d’anions o cations. La difusió en els sòlids i la conducció iònica en les sals sòlides impliquen forats i intersticials. Els semiconductors utilitzats en transistors són generalment semiconductors extrínsecs (o amb impureses) en contrast amb els semiconductors intrínsecs, en els quals la conductivitat elèctrica es deu principalment a defectes. Per exemple, l’addició d’una petita quantitat de P com a impuresa substitucional al Si augmenta significativament la seva conductivitat. Els àtoms de P tenen cinc electrons de valència en comparació amb els quatre del Si, cosa que produeix nivells d’energia electrònica addicionals situats lleugerament per sota de la banda de conducció, facilitant així l’excitació dels electrons cap a aquesta banda en el Si pur.
-
• Defectes de línea: Tenen a veure amb desplaçaments o alteracions d’una fila de posicions a la xarxa. Es poden provocar dislocacions d’aresta (de l’ordre de 10
 per cm
per cm en un metall templat o 10
en un metall templat o 10 per cm en un metall treballat en fred. Una dislocació de vora és un pla extra d’àtoms que s’estén parcialment a través del cristall, distorsionant així la seva estructura en els plans veïns i fent que el cristall sigui mecànicament feble. Una dislocació d’un tipus més complex és la dislocació de
cargol.
per cm en un metall treballat en fred. Una dislocació de vora és un pla extra d’àtoms que s’estén parcialment a través del cristall, distorsionant així la seva estructura en els plans veïns i fent que el cristall sigui mecànicament feble. Una dislocació d’un tipus més complex és la dislocació de
cargol.
-
• Defectes de pla: Bidimensionals. Els àtoms en l’extrem dels microcristalls poden ser més reactius per estar exposats amb més facilitat. Per exemple, un cristall d’empaquetament dens hexagonal pot contenir alguns plans on l’empaquetament és cúbic dens. La majoria dels sòlids cristal·lins no consisteixen en cristalls únics, sinó que estan formats per múltiples cristalls petits que es mantenen units. Els cristalls veïns tenen orientacions aleatòries, i les fronteres entre les seves cares són defectes de plans.
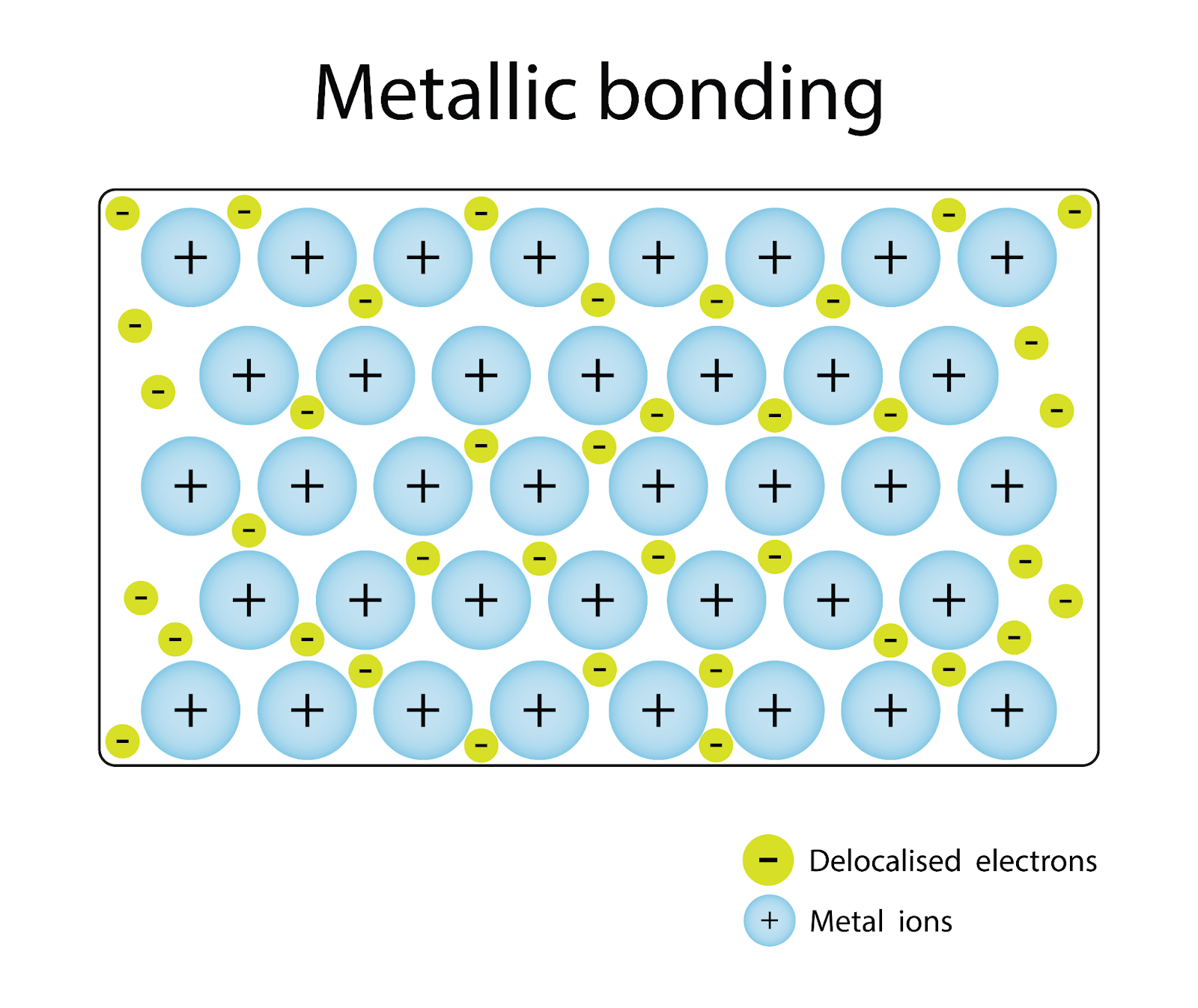
4.2.4 Enllaç metàl·lic
Els enllaços metàl·lics es formen quan un gran nombre d’àtoms comparteixen col·lectivament els seus electrons de valència. Aquesta situació és pròpia de metalls com el ferro, el coure o l’alumini, i explica per què aquests materials condueixen bé l’electricitat i la calor i, alhora, es poden deformar sense fracturar-se amb facilitat.
En un metall o en un aliatge, els electrons externs de cada àtom no queden lligats a un únic nucli, sinó que passen a formar part d’un núvol col·lectiu d’electrons lliures que recorre tota la xarxa cristal·lina. Tot i que els electrons es repel·leixen entre ells, l’atracció entre aquest núvol electrònic i els nuclis positius és suficient per mantenir cohesionada l’estructura.
El que distingeix els metalls de la resta de sòlids és que aquests electrons responen fàcilment als camps elèctrics aplicats, als gradients tèrmics i a la llum incident. D’aquí deriven les altes conductivitats elèctriques i tèrmiques, així com la seva gran reflectivitat òptica. Curiosament, propietats semblants també es
mantenen en metalls líquids, cosa que indica que el model d’electrons lliures continua sent útil fins i tot quan desapareix l’ordre cristal·lí. Les resistivitats elèctriques metàl·liques, habitualment entre  i
i  Ω cm, contrasten amb els valors molt més elevats de la resta de sòlids.
Ω cm, contrasten amb els valors molt més elevats de la resta de sòlids.
A més, el coeficient de temperatura de la resistivitat és positiu. Així doncs, els metalls es converteixen en conductors elèctrics més pobres a mesura que augmenta la temperatura. El contrari és cert per a totes les altres classes de sòlids. També, la conductivitat dels metalls purs es redueix sempre amb nivells baixos d’aliatge d’impuresa, un comportament contrari al d’altres sòlids. L’efecte tant de la temperatura com de l’addició d’elements d’aliatge sobre la conductivitat metàl·lica és augmentar la dispersió d’electrons, la qual cosa redueix el component net de moviment dels electrons en la direcció del camp elèctric aplicat.
Els electrons d’enllaç en els metalls no estan localitzats entre els àtoms i s’ha de dir que existeixen enllaços no direccionals. Això provoca que els àtoms llisquin uns sobre altres i es deformin plàsticament més fàcilment que no pas en el cas, per exemple, dels sòlids covalents que tenen enllaços atòmics direccionals.
4.3 Forces intermoleculars
Quan passem de descriure una molècula a descriure un líquid, un sòlid molecular o una dissolució, els enllaços interns ja no són l’únic factor rellevant. El comportament macroscòpic depèn sobretot de les forces que s’estableixen entre les molècules. Aquestes interaccions són més febles que els enllaços covalents o iònics, però governen propietats tan visibles com el punt d’ebullició, el punt de fusió o la solubilitat. En aquesta secció ens centrarem en dues famílies especialment importants: les forces de van der Waals i els ponts d’hidrogen.
4.3.1 Forces de van der Waals
Les forces de van der Waals agrupen diverses interaccions atractives que apareixen entre espècies neutres. Encara que cadascuna és feble, el seu efecte acumulat pot ser important, sobretot en molècules grans o polaritzables. Per això ajuden a explicar per què algunes substàncies condensen fàcilment, per què unes bullen abans que d’altres o per què dues espècies tenen més o menys afinitat quan es barregen.
En general, a distàncies molt curtes domina una repulsió forta, mentre que a distàncies una mica més grans apareix una atracció feble relacionada amb la polarització electrònica. Aquest compromís entre repulsió i atracció s’il·lustra sovint amb el potencial de Lennard-Jones 12/6:
![(4.3.1) \begin{equation} V(r)=4\varepsilon \left [ \left (\frac {\sigma }{r}\right )^{12} - \left (\frac {\sigma }{r}\right )^6 \right ] \end{equation}](web_GEAForcesInt-images/image-131.svg)
on  és la distància intermolecular,
és la distància intermolecular,  fixa la profunditat del pou de potencial i
fixa la profunditat del pou de potencial i  és una distància característica. El terme
és una distància característica. El terme  representa la repulsió de curt abast i el terme
representa la repulsió de curt abast i el terme  l’atracció de dispersió. Tot i que una interacció individual és feble, moltes d’elles poden sumar-se i afectar de manera clara les propietats d’un líquid o d’un sòlid molecular.
l’atracció de dispersió. Tot i que una interacció individual és feble, moltes d’elles poden sumar-se i afectar de manera clara les propietats d’un líquid o d’un sòlid molecular.
4.3.1.1 Forces de dispersió de London
Les interaccions atractives són universals. Les molècules no han de tenir càrrega neta ni una asimetria permanent de càrregues perquè apareguin forces intermoleculars.
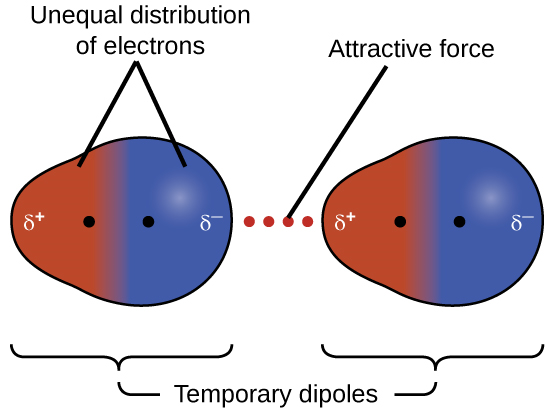
Les molècules no polars poden interactuar mitjançant les forces de dispersió de London. Aquestes forces apareixen perquè la distribució electrònica fluctua contínuament: un dipol instantani en una molècula pot
induir-ne un altre en una molècula veïna. En el cas més simple, el moment dipolar induït és proporcional al camp elèctric aplicat, i la polaritzabilitat  és una constant que depèn de la molècula[4]:
és una constant que depèn de la molècula[4]:

Aquest procés crea una atracció que tendeix a ser més intensa com més grans i més deformables són els núvols electrònics. Les forces de London expliquen, per exemple, la cohesió entre espècies no polars com l’argó ( ) o el metà (
) o el metà ( ). La Figura 4.7 mostra l’origen instantani i induït d’aquesta atracció.
). La Figura 4.7 mostra l’origen instantani i induït d’aquesta atracció.
-
• Totes les espècies químiques, cations i anions, tenen una determinada capacitat de polarització i presenten una polaritzabilitat característica.
-
• Els cations polaritzen els anions:
-
1. La capacitat de polarització dels cations serà més gran com més gran sigui la seva relació càrrega/radi.
-
2. La polaritzabilitat dels anions serà més gran com més gran sigui el seu radi i la seva càrrega.
-
3. A igualtat de relació càrrega/radi, la capacitat de polarització d’un metall de transició és més gran que la d’un metall dels grups principals.
-
4.3.1.2 Forces dipol-dipol
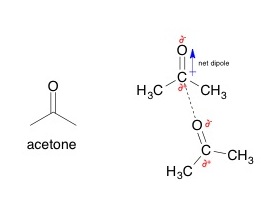
Les forces dipol-dipol són atraccions entre molècules polars amb moments dipolars permanents. En general, com més polar és una molècula, més important pot ser aquesta contribució. Són rellevants en substàncies com l’aigua ( ) o l’amoníac (
) o l’amoníac ( ). La Figura 4.8 il·lustra aquesta orientació entre molècules d’acetona.
). La Figura 4.8 il·lustra aquesta orientació entre molècules d’acetona.
4.3.2 Ponts d’hidrogen
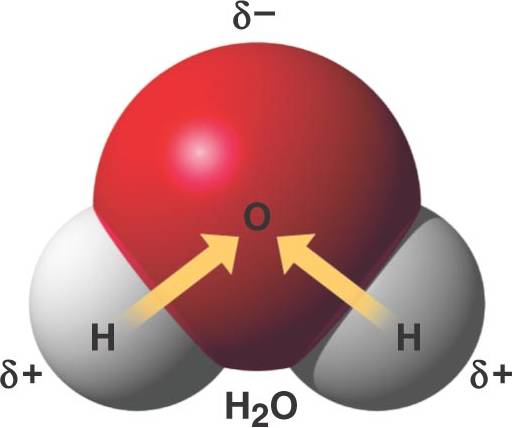
Els ponts d’hidrogen apareixen quan un hidrogen unit a un àtom molt electronegatiu, com O, N o F, interactua amb un altre centre ric en densitat electrònica. Són més intensos i més direccionals que la majoria de forces de van der Waals, i per això tenen conseqüències molt visibles en substàncies com l’aigua, l’amoníac o molts compostos orgànics oxigenats i nitrogenats. La Figura 4.9 mostra el cas senzill de dues molècules d’aigua.
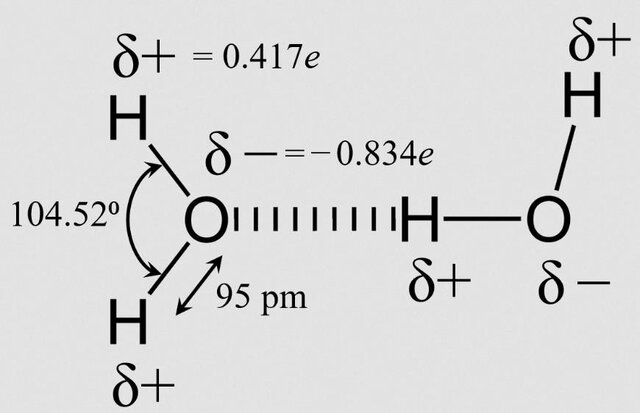
Les distàncies d’enllaç entre els àtoms d’hidrogen i oxigen en un pont d’hidrogen són més curtes que les distàncies de van der Waals, però més llargues que les distàncies covalents. Els ponts d’hidrogen són diferents de les forces de van der Waals perquè són més forts i tenen una direccionalitat més marcada. La clau de la diferència és que els ponts d’hidrogen, típicament, tenen un cert caràcter covalent i una penetració mútua d’electrons dins del radi de van der Waals dels àtoms. Segons la IUPAC (International Union of Pure and Applied Chemistry), un pont d’hidrogen és una interacció atractiva en la que:
-
• l’àtom d’hidrogen en el grup donador
 és més electropositiu que
és més electropositiu que  , i
, i
-
• les forces fisiques involucrades han d’incloure interaccions electrostàtiques, és a dir, no poden ser únicament forces de dispersió.
4.4 Líquids i dissolucions
4.4.1 Teoria cinètica
La teoria cinètico-molecular també es pot aplicar als líquids, però amb una diferència important respecte dels gasos: en un líquid les partícules són molt més properes i les forces intermoleculars ja no es poden ignorar. Això fa que un líquid tingui volum gairebé constant i sigui poc compressible, però al mateix temps mantingui la capacitat de fluir.
Des d’un punt de vista microscòpic, les molècules d’un líquid es mouen contínuament, xoquen i es reorganitzen sense parar. No estan fixades com en un sòlid, però tampoc es mouen gairebé lliures com en un gas. Per això un líquid combina desordre i mobilitat amb una cohesió apreciable entre partícules.
Una manifestació visible d’aquest moviment és el moviment Brownià. Robert Brown va observar que partícules molt petites suspeses en un fluid segueixen una trajectòria irregular i aparentment caòtica. Això passa perquè reben impactes continus i desiguals de les molècules del medi. Quan la mida de la partícula és molt petita, de l’ordre de 1 · 10−6 m, l’efecte acumulat d’aquests xocs fa que el moviment observat sembli completament aleatori. La Figura 4.10 en mostra una representació esquemàtica.
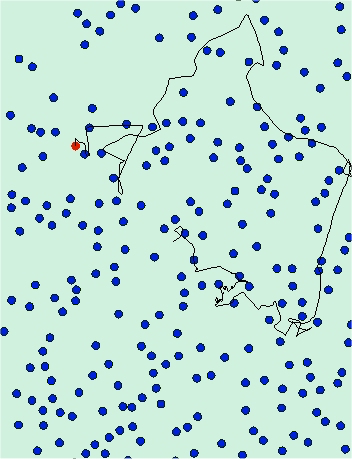
El moviment Brownià es pot descriure amb un model estocàstic en què cada canvi de posició és aleatori i s’extreu d’una distribució normal de mitjana  i variància
i variància  , és a dir,
, és a dir,  . En altres paraules, com més temps observem la partícula, més s’eixampla el conjunt de posicions possibles.
. En altres paraules, com més temps observem la partícula, més s’eixampla el conjunt de posicions possibles.
Exercici 3. Moviment Brownià
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Usant R, prova d’executar aquest script que mostra com simular el moviment Brownià d’una partícula en un líquid (extret de http://www.phytools.org/eqg/phytools/):
1 t <− 0:100 # temps de simulacio 2 sig2 <− 0.01 3 ## primer , calcula un conjunt de desviacions aleatòries puntuals 4 x <− rnorm ( n = length ( t ) − 1, sd = sqrt ( sig2 ) ) 5 ## després , acumula'n els resultats 6 x <− c (0, cumsum(x) ) 7 plot ( t , x , type = ” l ” , ylim = c (−2, 2) )
Després, executa el següent script, que produeix 10000 simulacions diferents:
1 nsim <− 1000 2 ## creo una matriu que hostatgi totes les simulacions 3 X <− matrix (0, nsim , length ( t ) ) 4 for ( i in 1: nsim ) X[ i , ] <− c (0, cumsum(rnorm(n = length ( t ) − 1, sd = sqrt ( sig2 ) ) ) ) 5 plot ( t , X [1, ], xlab = ” temps ” , ylab = ” desviacions ” , ylim = c (−2, 2) , type = ” l ” ) 6 for ( i in 1: nsim ) lines ( t , X[ i , ])
Exercici 4. Variança
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Per saber la variança que s’obté de la simulació podem fer
1 var ( X [, length ( t ) ])
i per mostrar l’histograma de posicions finals:
1 hist ( X [, length ( t ) ])
o bé:
1 plot ( density ( X [, length ( t ) ]) )
Calcula la variança de la distribució per a diferents valors del nombre de simulacions o el temps simulat.
A partir de la teoria cinètico-molecular podem entendre que la temperatura continua mesurant l’agitació tèrmica mitjana de les partícules. Això ajuda a interpretar per què, en escalfar un líquid, augmenten la mobilitat molecular, la difusió i la facilitat amb què algunes molècules poden escapar de la superfície.
4.4.2 Equilibris de fase
Quan un líquid passa a vapor, parlem de vaporització. Aquest procés pot tenir lloc només a la superfície, cas en què rep el nom d’evaporació, o bé a tot el volum del líquid, cas en què tenim ebullició. En tots dos casos cal aportar energia per vèncer les forces intermoleculars, de manera que es tracta d’un procés endotèrmic. El procés invers és la condensació.
Exercici 5. Equilibri dinàmic i saturació
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Explica, segons la teoria cinètico-molecular, la Figura 4.11. Com interpretes els termes equilibri dinàmic i saturació?
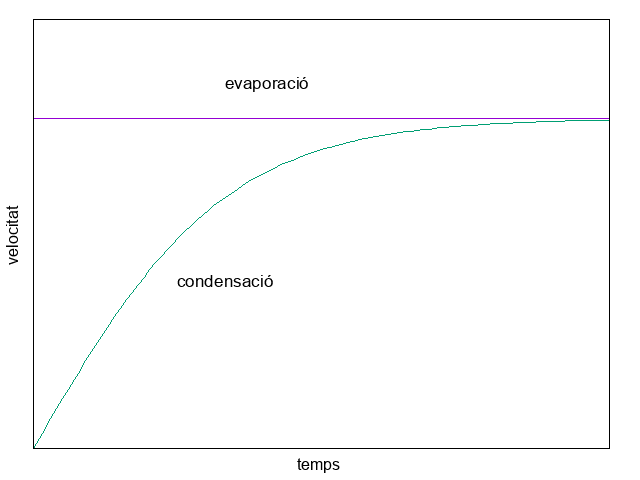
Quan un líquid i el seu vapor arriben a l’equilibri dins d’un recipient tancat, el vapor exerceix una pressió característica. Aquesta magnitud és la pressió de vapor,  , i depèn de la temperatura. A mesura que la temperatura augmenta, més molècules tenen prou energia per escapar de la fase líquida i, per tant, la pressió de vapor també creix, tal com mostra la Figura 4.12.
, i depèn de la temperatura. A mesura que la temperatura augmenta, més molècules tenen prou energia per escapar de la fase líquida i, per tant, la pressió de vapor també creix, tal com mostra la Figura 4.12.
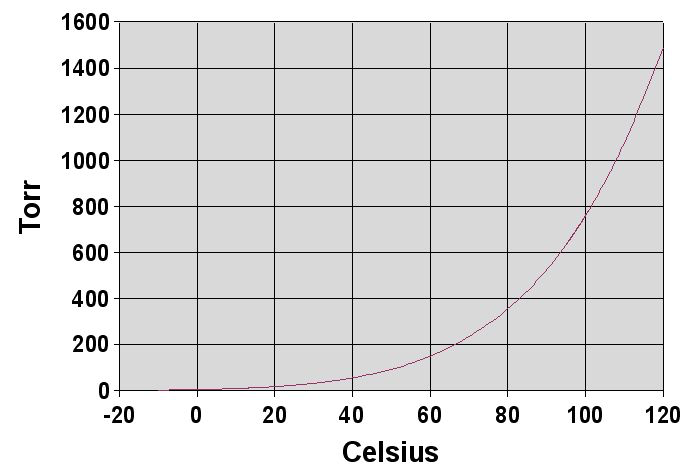
La temperatura d’ebullició normal és la temperatura a la qual un líquid bull quan la pressió externa és d’1 atm. En general, la temperatura d’ebullició no és una propietat absoluta del líquid: depèn tant de la naturalesa de la substància com de la pressió exterior que se li imposa.
La relació entre pressió de vapor, temperatura d’ebullició i entalpia de vaporització és molt directa (vegeu la Taula 4.5). Si les forces intermoleculars són intenses, costa més separar les molècules, i això es tradueix en valors més elevats de  i de
i de  . Aquest resultat fa referència encara a substàncies pures; més endavant veurem com la presència d’un solut modifica aquest equilibri.
. Aquest resultat fa referència encara a substàncies pures; més endavant veurem com la presència d’un solut modifica aquest equilibri.
| Líquid | Naturalesa | / 105 Pa |
/ °C |
/ kJ/mol |
 |
no polar |  |
-268.9 | 0.1003 |
|
no polar | |
-252.7 | 0.9028 |
|
no polar | |
-161.4 | 9.263 |
 |
no polar | 2.03 | -1.5 | 24.24 |
 |
no polar | 0.121 | 76.7 | 34.57 |
|
polar | 10.1 | -33.6 | 20.15 |
|
polar | 0.0233 | 100.0 | 40.62 |
 |
polar | 0.0586 | 78.5 | 40.44 |
 |
polar | 5.06 | -23.7 | 22.61 |
 |
polar | 0.247 | 56.5 | 31.94 |
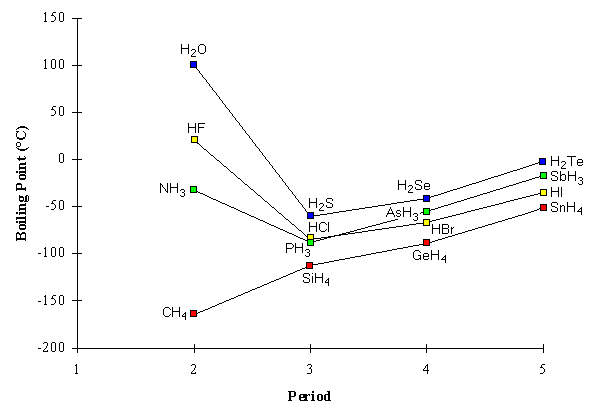
La Figura 4.13 mostra com la evoluciona dins d’una mateixa família de la taula periòdica, i també posa de manifest que alguns hidrurs se n’aparten clarament perquè poden establir ponts d’hidrogen.
4.4.3 Propietats crítiques
L’estudi experimental dels gasos va mostrar ben aviat que no tots es deixaven liquar amb la mateixa facilitat. Faraday va aconseguir liquar clor el 1823, però gasos com , o van requerir tècniques més sofisticades. Quan Thomas Andrews va estudiar el  , va veure que la liqüefacció només era possible per sota d’una certa temperatura. D’aquí neix el concepte de temperatura crítica,
, va veure que la liqüefacció només era possible per sota d’una certa temperatura. D’aquí neix el concepte de temperatura crítica,  : per sobre d’aquest valor, un gas ja no es pot liquar només augmentant la pressió (Figura 4.14).
: per sobre d’aquest valor, un gas ja no es pot liquar només augmentant la pressió (Figura 4.14).
En el punt crític, la pressió crítica  coincideix amb la pressió de vapor del líquid a . És el punt final de la corba líquid-vapor: a temperatures superiors ja no podem distingir clarament entre líquid i gas.
coincideix amb la pressió de vapor del líquid a . És el punt final de la corba líquid-vapor: a temperatures superiors ja no podem distingir clarament entre líquid i gas.
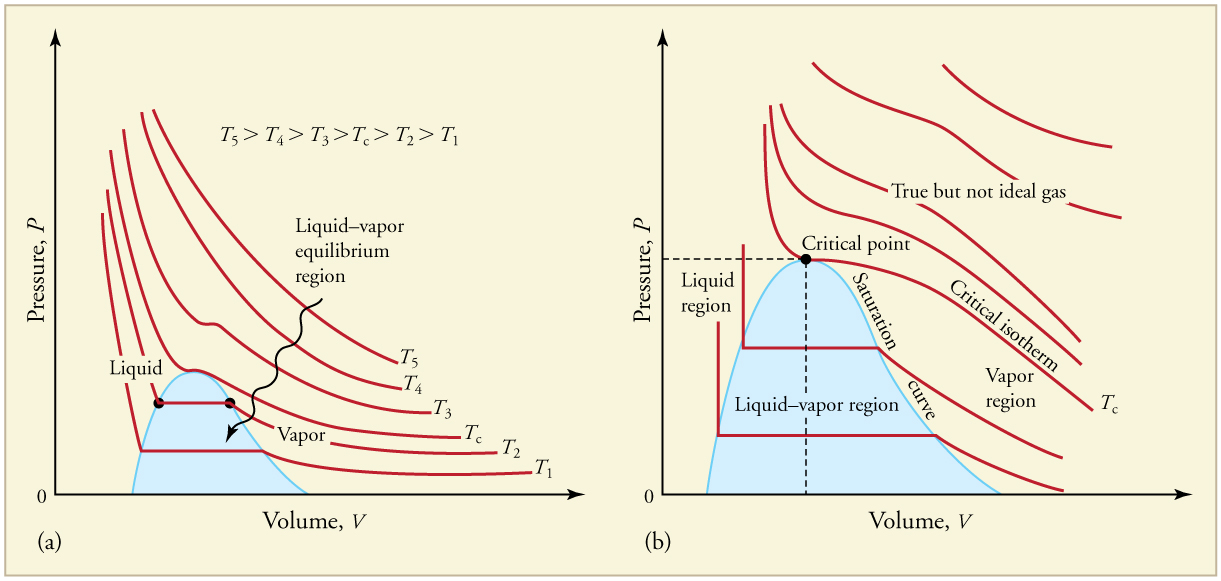
 i
i  a una temperatura donada. Les corbes inferiors deixen de ser hipèrboles perquè el gas es torna no ideal; b) el terme “vapor” es refereix a gas a una temperatura inferior al punt d’ebullició. Veure valors de punts crítics de substàncies comunes a http://philschatz.com/physics-book/contents/m42218.html.Font: [14]
a una temperatura donada. Les corbes inferiors deixen de ser hipèrboles perquè el gas es torna no ideal; b) el terme “vapor” es refereix a gas a una temperatura inferior al punt d’ebullició. Veure valors de punts crítics de substàncies comunes a http://philschatz.com/physics-book/contents/m42218.html.Font: [14]
La Figura 4.15 mostra, de manera simplificada, les regions de fase de l’aigua i la posició qualitativa del punt triple i del punt crític.

En el punt crític, la densitat i la resta de propietats macroscòpiques del líquid i del gas convergeixen fins a fer-se indistingibles.
Exercici 6. Què crema més?
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Què produirà una cremada més greu: una massa  d’(g) a 100 °C o la mateixa massa d’aigua líquida a la mateixa temperatura?
d’(g) a 100 °C o la mateixa massa d’aigua líquida a la mateixa temperatura?
Exercici 7. Ebullició aigua
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
En un recipient hi ha aigua líquida. Es connecta el recipient a una bomba de buit i es va abaixant la pressió sobre el líquid. Si la temperatura és de 60 °C, a quina pressió bullirà l’aigua?
4.4.4 Dissolucions i solubilitats
Una dissolució és una mescla homogènia la composició de la qual pot variar de manera contínua dins d’uns certs límits. Aquesta definició és útil per començar, tot i que més endavant veurem casos fronterers, com ara el sabó en aigua, en què la distinció entre dissolució i altres sistemes dispersos es torna menys nítida.
Per entendre per què una substància es dissol o no en una altra, no n’hi ha prou amb mirar només si l’energia disminueix. En els processos moleculars hi ha dues tendències generals: la d’assolir configuracions energèticament favorables i la d’augmentar el desordre accessible al sistema. Aquest balanç queda resumit en l’energia lliure de Gibbs:
![\[ \Delta G=\Delta H-T\Delta S \]](web_GEAForcesInt-images/image-179.svg)
Quan  , el procés és espontani; quan
, el procés és espontani; quan  , el sistema és en equilibri. Aquesta idea serà especialment útil per interpretar la formació de dissolucions ideals i no ideals, així com la solubilitat de sòlids i gasos.
, el sistema és en equilibri. Aquesta idea serà especialment útil per interpretar la formació de dissolucions ideals i no ideals, així com la solubilitat de sòlids i gasos.
Per descriure quantitativament una dissolució necessitem una mesura de concentració. Les més habituals són:
-
• Fracció molar:
 i
i  .
.
-
• Molalitat: nombre de mols de solut per 1 kg de dissolvent.
-
• Molaritat: nombre de mols per 1 L de dissolució.
-
• Normalitat: nombre de pesos equivalents-gram de solut per 1 L de dissolució.
La solubilitat és la quantitat màxima de solut que pot romandre dissolta en un dissolvent a una temperatura i una pressió determinades. En altres paraules, és la concentració del solut en una dissolució saturada. Aquesta magnitud és fonamental perquè condiciona la separació de components, l’estabilitat de formulacions i el comportament de molts processos químics i industrials.
La solubilitat depèn de la naturalesa química del solut i del dissolvent, però també de variables externes com la temperatura i la pressió. En general, la solubilitat de la majoria de sòlids augmenta quan ho fa la temperatura, mentre que en molts gasos passa just el contrari. La pressió, en canvi, acostuma a ser especialment rellevant en la solubilitat dels gasos.
Per entendre l’efecte de la temperatura sobre la solubilitat, ens serà útil el principi de Le Chatelier: quan un sistema en equilibri pateix una pertorbació, evoluciona en el sentit que tendeix a contrarestar-la.
Exercici 8. Efecte de  en la solubilitat
en la solubilitat
Raona perquè per a una dissolució en la qual  , un augment de la temperatura fa que la solubilitat disminueixi, i a l’inrevés.
, un augment de la temperatura fa que la solubilitat disminueixi, i a l’inrevés.
4.4.4.1 Solucions ideals i no ideals
En una dissolució ideal, les interaccions solut-dissolvent són comparables a les que ja existien entre les espècies pures. Per això la barreja es forma sense un intercanvi apreciable de calor i la pressió de vapor segueix la llei de Raoult:

L’equació 4.4.1 informa, doncs, sobre com varia la pressió de vapor amb la composició. A partir d’aquesta idea, i només per a dissolucions prou diluïdes, es pot deduir una segona expressió que ja no dona la pressió de vapor, sinó directament el desplaçament del punt d’ebullició:

on  és la constant ebullioscòpica del dissolvent. En altres paraules: la llei de Raoult és la relació fonamental, mentre que l’equació 4.4.5 n’és una conseqüència pràctica per calcular l’elevació ebullioscòpica.
és la constant ebullioscòpica del dissolvent. En altres paraules: la llei de Raoult és la relació fonamental, mentre que l’equació 4.4.5 n’és una conseqüència pràctica per calcular l’elevació ebullioscòpica.
|
Taula 4.6: Temperatura d’ebullició i constant ebulloscòpica de diversos dissolvents.Font: [2] |
|||
| Dissolvent | Fórmula molecular |  (°C) (°C) |
 (°C kg/mol) (°C kg/mol) |
| Dissolvent | Fórmula molecular | (°C) |
(°C kg/mol) |
| Aigua | |
100.0 | 0.51 |
| Etanol |  |
78.4 | 1.22 |
| Benzè |  |
80.1 | 2.53 |
| Éter etílic |  |
34.6 | 2.02 |
| Cloroform |  |
64.3 | 3.63 |
De manera anàloga, per al punt de congelació obtenim:

Cal recordar que, si hi ha més d’un solut, hem de tenir en compte la molalitat total. I si la dissolució conté més d’un component volàtil, la llei de Raoult s’ha d’aplicar a cadascun d’ells i sumar-ne les pressions parcials. Per a dos components volàtils, per exemple:

En una dissolució ideal binària, el vapor queda enriquit en el component més volàtil. Aquesta idea es visualitza bé amb diagrames com els de la Figura 4.16: a partir de la diferència de volatilitat entre dos components podem seguir tant la composició del vapor com l’efecte de successives etapes de destil·lació fraccionada.
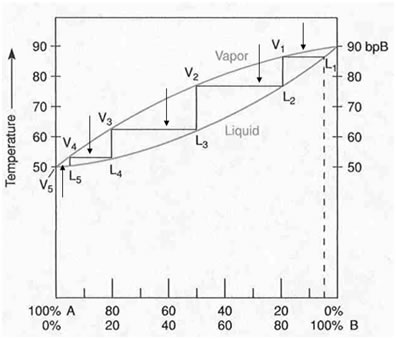
 d’ebullició donada, els punts de tall de les dues corbes a aquesta . b) Destil·lació fraccionada d’una disslució ideal.
d’ebullició donada, els punts de tall de les dues corbes a aquesta . b) Destil·lació fraccionada d’una disslució ideal.
En una dissolució ideal no hi ha un efecte entàlpic net de barreja ( ); la força motriu és essencialment entròpica i, per això, el procés de mescla és espontani sempre que
); la força motriu és essencialment entròpica i, per això, el procés de mescla és espontani sempre que  .
.
Quan les interaccions entre espècies diferents no són equivalents a les de les substàncies pures, apareixen desviacions respecte de la llei de Raoult. Aquestes desviacions poden ser positives o negatives (vegeu la Figura 4.17).
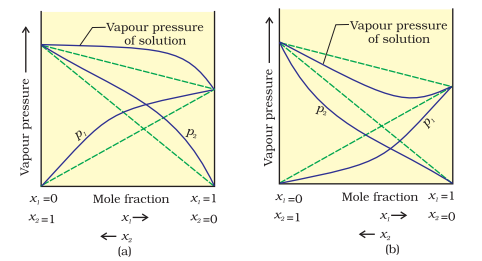
Des del punt de vista energètic, és útil distingir dos casos:
-
• La dissolució absorbeix calor en formar-se (
 ). Això sol passar quan es barregen substàncies amb interaccions mútues menys favorables que les que tenien per separat, com en moltes mescles de compostos polars amb no polars. Un exemple és la barreja d’acetona amb disulfur de carboni (
). Això sol passar quan es barregen substàncies amb interaccions mútues menys favorables que les que tenien per separat, com en moltes mescles de compostos polars amb no polars. Un exemple és la barreja d’acetona amb disulfur de carboni ( ), associada a una desviació positiva.
), associada a una desviació positiva.
-
• La dissolució desprèn calor en formar-se (
 ). Això ocorre quan en la barreja apareixen interaccions especialment favorables. Per exemple, en barrejar cloroform () i acetona (
). Això ocorre quan en la barreja apareixen interaccions especialment favorables. Per exemple, en barrejar cloroform () i acetona ( ) es poden establir interaccions específiques que estabilitzen la dissolució i donen lloc a una desviació negativa. La Figura 4.18 mostra una interacció possible entre aquestes dues molècules.
) es poden establir interaccions específiques que estabilitzen la dissolució i donen lloc a una desviació negativa. La Figura 4.18 mostra una interacció possible entre aquestes dues molècules.
En aquest segon cas, la pressió de vapor és menor que la prevista per la llei de Raoult (Figura 4.17b).

Per tant, si en formar-se una dissolució observem un canvi de temperatura apreciable, és un bon indici que la barreja no és ideal. Ara bé, en el límit de dilució infinita el dissolvent acostuma a recuperar un comportament proper a l’ideal. En dissolucions no ideals binàries, a més, ja no és cert que el vapor quedi sempre enriquit en el component més volàtil.
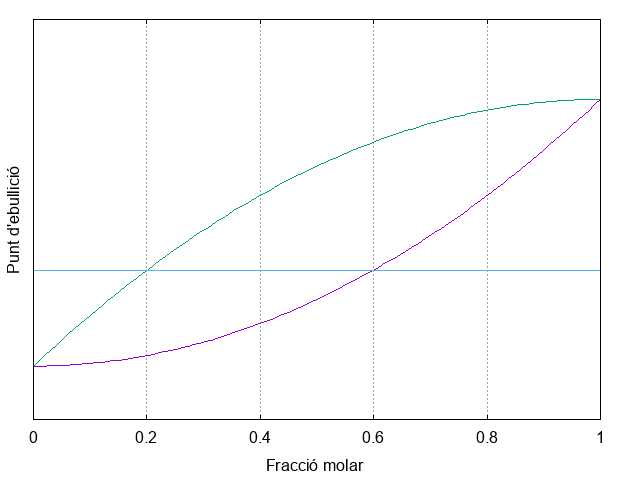
Les dissolucions que destil·len sense canviar de composició s’anomenen azeotròpiques. Per exemple, si fem bullir durant prou temps una barreja d’aigua i àcid clorhídric, la composició evoluciona fins a una dissolució amb un 20,22 % en massa d’.
Exercici 9. Azeòtrops
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Un azeòtrop positiu prové d’una desviació també positiva de la llei de Raoult. a) Dibuixa la corba de Temperatura d’ebullició vs composició per a un azeòtrop positiu basant-te en les Figures 4.16a i 4.17a. b) Raona el resultat de fer una destil·lació a partir de diverses composicions d’aquesta mescla. c) què succeiria en un azeòtrop negatiu?
La Figura 4.19 mostra un exemple d’azeòtrop en una mescla binària.
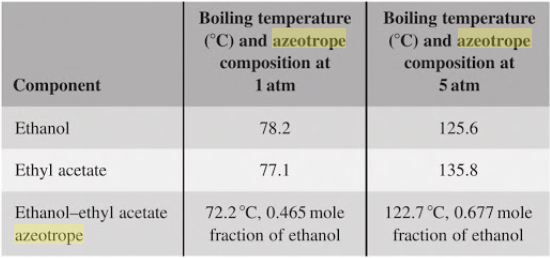
Exercici 10. Barreges equimolars
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Volem separar una barreja equimolar d’etanol i acetat etílic per destil·lació en productes relativament purs. La barreja forma un azeòtrop de mínim punt d’ebullició segons la Figura 4.19. No obstant, la composició de l’azeòtrop és sensible a la pressió, mostrant un increment significatiu de la fracció molar de l’etanol quan incrementa la pressió, com es mostra a la Figura. Dibuixa un esquema aproximat per a la separació de les dues components de la barreja que tregui profit d’aquest fet.
4.4.4.2 Propietats colligatives
En dissolucions diluïdes de soluts no volàtils, la presència de partícules de solut redueix l’activitat del dissolvent i, per tant, la seva pressió de vapor. Aquesta és la idea de fons que explica les propietats colligatives. Per a l’elevació del punt d’ebullició, la conseqüència pràctica és:

on és la constant ebullioscòpica del dissolvent i és la molalitat.

En presència d’electròlits, només cal incorporar el factor de van’t Hoff:

on:
-
•
 és el factor de van’t Hoff, que val 1 per a soluts no dissociats i coincideix amb el nombre total d’ions generats si el solut es dissocia completament,
és el factor de van’t Hoff, que val 1 per a soluts no dissociats i coincideix amb el nombre total d’ions generats si el solut es dissocia completament,
-
•
és la constant ebullioscòpica del dissolvent (per a l’aigua,  ), i
), i
-
•
és la molalitat, calculada com:

Activitat i concentració
En dissolucions molt diluïdes, sovint podem identificar activitat i concentració sense cometre un gran error. En dissolucions concentrades, però, les interaccions entre partícules fan que la concentració deixi de descriure completament el comportament real del sistema. En aquests casos, la magnitud termodinàmica rellevant és l’activitat.
EXEMPLE 1. Exemple amb no-electròlit
Dissoldre 75,0 g de glicerina ( ) en 240,0 g d’aigua.
) en 240,0 g d’aigua.

Punt d’ebullició de la solució:
![\[ T_b = \qty {100}{\celsius } + \qty {1.74}{\celsius } = \qty {101.74}{\celsius } \]](web_GEAForcesInt-images/image-223.svg)
EXEMPLE 2. Exemple amb electròlit
Dissoldre 5,4 g de  en 36,0 g d’aigua.
en 36,0 g d’aigua.
![\[ \ch {FeCl3 (aq) -> Fe^{3+} (aq) + 3Cl^{-} (aq)} \Rightarrow \text {4 ions} \]](web_GEAForcesInt-images/image-225.svg)

Punt d’ebullició de la solució:
![\[ T_b = \qty {100}{\celsius } + \qty {1.89}{\celsius } = \qty {101.89}{\celsius } \approx \qty {102}{\celsius } \]](web_GEAForcesInt-images/image-227.svg)
4.4.4.3 Diagrama de fases d’una dissolució
Els efectes colligatius es veuen especialment bé quan comparem el diagrama de fases d’un dissolvent pur amb el d’una dissolució preparada a partir d’aquest dissolvent. La Figura 4.20 resumeix en una sola imatge la disminució de la pressió de vapor, l’elevació del punt d’ebullició i la depressió del punt de congelació.
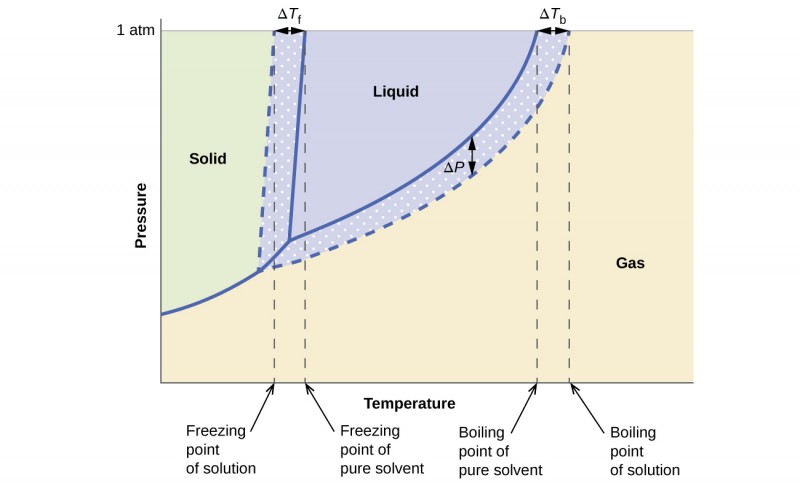
El gràfic mostra, per a una mateixa pressió, que la dissolució congela a una temperatura més baixa i bull a una temperatura més alta que el dissolvent pur. Alhora, per a una mateixa temperatura, la seva pressió de vapor és menor. Aquestes diferències apareixen representades com  ,
,  i
i  .
.
La corba líquid-vapor de la dissolució queda per sota de la del dissolvent pur: això és la disminució de la pressió de vapor, . Com a conseqüència, per arribar a una pressió externa donada cal escalfar més la dissolució, i el punt d’ebullició s’eleva en .
La corba sòlid-líquid, en canvi, es desplaça cap a temperatures més baixes, reflectint la depressió del punt de congelació, . En molts casos, quan la dissolució congela és el dissolvent qui forma sòlid pur i deixa el solut exclòs de la fase sòlida. Per això les transicions que impliquen només el dissolvent en fase sòlida o gasosa queden molt menys afectades pels efectes colligatius.
Exercici 11. Diagrama de fases d’aigua i solució aquosa
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Com seria el diagrama de fases de l’aigua, comparat amb el de la Figura 4.20? Spoiler.
4.4.4.4 Solubilitat de Sals
La solubilitat de les sals en aigua depèn del balanç entre l’energia de xarxa del cristall i l’estabilització que proporciona la hidratació dels ions. En termes generals, ions petits i molt carregats tendeixen a formar sòlids més estables i, sovint, menys solubles. La temperatura pot modificar aquest equilibri de manera important, mentre que la pressió sol tenir un efecte petit sobre la solubilitat de sòlids iònics.
4.4.4.5 Solubilitat de Gasos
En el cas dels gasos, la situació és diferent. La seva solubilitat en un líquid depèn fortament de la pressió parcial del gas i, en molts casos, disminueix quan la temperatura augmenta. Aquesta dependència es resumeix en la llei de Henry1, que relaciona la concentració dissolta amb la pressió parcial del gas sobre el líquid:

on:
-
•
 és la concentració del gas dissolt en el líquid,
és la concentració del gas dissolt en el líquid,
-
•
 és la constant de Henry, que depèn de la naturalesa del gas i del líquid(veure Taula 4.7), així com de la temperatura, i
és la constant de Henry, que depèn de la naturalesa del gas i del líquid(veure Taula 4.7), així com de la temperatura, i
-
•
és la pressió parcial del gas sobre el líquid.
La llei de Henry ens diu, doncs, que si augmentem la pressió parcial d’un gas, la seva solubilitat en el líquid també augmenta. És una aproximació especialment útil per a gasos poc solubles i en condicions relativament suaus.
4.5 Controlant la temperatura
4.5.1 Introducció
En un automòbil, una part molt important de l’energia acaba convertida en calor. De vegades això és intencionat, com en la frenada per fricció; en altres casos és una conseqüència inevitable del funcionament dels components, com passa en el motor de combustió. Encara que hi hagi sistemes de recuperació energètica, cap no elimina del tot la necessitat de gestionar aquesta calor.
Si aquesta calor no s’evacua bé, apareixen deformacions, pèrdua d’eficiència o fins i tot fallades estructurals. A més, el control tèrmic no afecta només el motor: també condiciona el confort de l’habitacle i la fiabilitat general del vehicle. Per això, la química dels refrigerants, dels materials de fricció i dels sistemes d’intercanvi tèrmic és una peça central del disseny automobilístic.
4.5.2 El refrigerant del motor
El refrigerant del motor d’un automòbil és, principalment, una mescla d’aigua i etilenglicol, amb additius anticorrosius, colorants i altres components auxiliars. La seva utilitat es basa justament en els dos efectes colligatius que hem vist: l’elevació del punt d’ebullició i la depressió del punt de congelació.
Tal com hem vist abans, perquè una substància es dissolgui bé cal que pugui establir interaccions intermoleculars favorables amb el dissolvent. En l’aigua, la interacció dominant és el pont d’hidrogen. L’etilenglicol ( ) conté dos grups
) conté dos grups  i, per tant, pot integrar-se fàcilment en la xarxa de ponts d’hidrogen de l’aigua; això n’explica la gran solubilitat. La Figura 4.21 representa aquests ponts d’hidrogen.
i, per tant, pot integrar-se fàcilment en la xarxa de ponts d’hidrogen de l’aigua; això n’explica la gran solubilitat. La Figura 4.21 representa aquests ponts d’hidrogen.
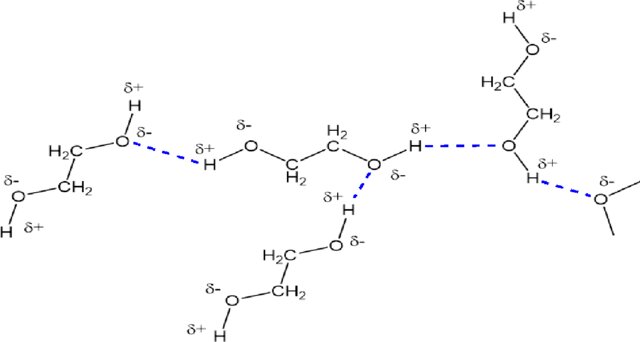
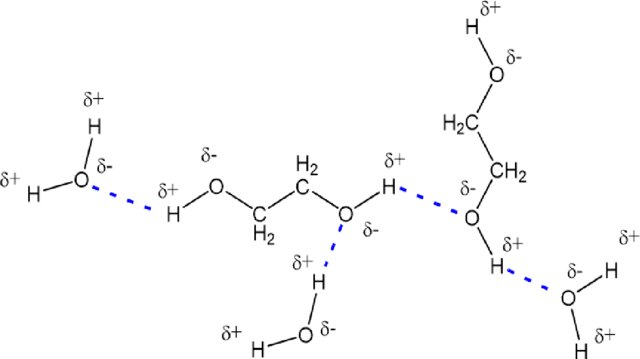
Un refrigerant típic pot contenir aproximadament un 50 % en massa d’aigua i un 50 % en massa d’etilenglicol. És, per tant, una dissolució molt concentrada si la comparem amb els exemples ideals i diluïts de la química general. En rigor caldria treballar amb activitats, però per entendre el mecanisme qualitatiu podem aproximar el sistema com si fos ideal.
Per estimar el punt d’ebullició del refrigerant, primer cal saber a quina temperatura bull l’aigua a la pressió del circuit i, després, sumar-hi l’elevació colligativa deguda a l’etilenglicol. A una pressió de 2 atm (aproximadament 30 psi), l’aigua bull cap a 121 °C. Això ja mostra que la pressurització del sistema és clau per ampliar la finestra de funcionament del líquid refrigerant.






Com s’evita, doncs, la transició de fase de l’aigua dins del circuit?
-
• El sistema de refrigeració està pressuritzat, cosa que eleva el punt d’ebullició (fins a 121 °C sota 2 atm) (veure Exercici 40).
-
• L’aigua es barreja amb etilenglicol (
 ), aprofitant les propietats colligatives: elevació del punt d’ebullició i depressió del punt de congelació.
), aprofitant les propietats colligatives: elevació del punt d’ebullició i depressió del punt de congelació.
Les propietats colligatives depenen sobretot del nombre de partícules de solut, no de la seva identitat química. En el cas del refrigerant, però, cal recordar que un 50 % en massa d’etilenglicol continua sent una dissolució molt concentrada i que, per tant, el càlcul anterior és només una aproximació útil.
4.5.3 Sal de carretera
Sals com  i
i  són higroscòpiques: capten aigua de l’aire i, en hidratar-se, alliberen calor. Aquesta calor pot ajudar a fondre el gel ja existent. A més, la dissolució salina que es forma té un punt de congelació més baix que l’aigua pura, de manera que dificulta la formació de nou gel.
són higroscòpiques: capten aigua de l’aire i, en hidratar-se, alliberen calor. Aquesta calor pot ajudar a fondre el gel ja existent. A més, la dissolució salina que es forma té un punt de congelació més baix que l’aigua pura, de manera que dificulta la formació de nou gel.
El procés és termodinàmicament afavorit ( ), amb guany d’entropia i alliberament d’energia lliure durant la hidratació. Veure l’Exercici 44.
), amb guany d’entropia i alliberament d’energia lliure durant la hidratació. Veure l’Exercici 44.
4.5.4 Controvèrsia sobre els refrigerants
La substitució dels clorofluorocarbons (CFC) va reduir de manera important l’impacte sobre la capa d’ozó, però no va eliminar el problema ambiental dels refrigerants. Alguns compostos alternatius, com el R-134a, presenten un potencial d’escalfament global molt elevat.
El R-134a (1,1,1,2-tetrafluoroetà) és un hidrofluorocarbur popular com a refrigerant en sistemes de refrigeració i aire condicionat, inclosos molts sistemes d’automòbils. Té un alt potencial d’escalfament global (GWP, global warming potential) de 1430. No obstant això, el seu ús es veu afectat pel Reglament (UE) 517/2014, que impulsa una reducció progressiva dels gasos fluorats amb alt GWP abans del 2030. Aquesta regulació ha provocat una escassetat important de R-134a i un augment de preus de fins a 400 %, ja que els fabricants passen a refrigerants més ecològics.
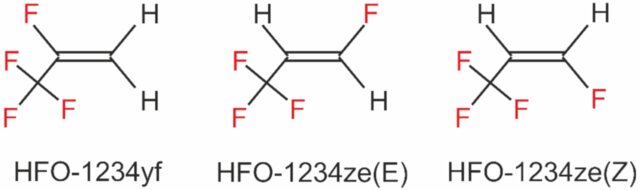
S’han desenvolupat alternatives com el R-1234yf i el R-1234ze(E), hidrofluoroolefines (HFO) amb valors de GWP molt baixos. El R-1234yf té un GWP de 4 i un rendiment similar al del R-134a, però presenta riscos d’inflamabilitat que obliguen a modificar els sistemes per garantir-ne la seguretat. El R-1234ze(E) té característiques semblants i s’utilitza especialment en grans sistemes d’aire condicionat. Malgrat les opcions existents, no tots els refrigerants alternatius compleixen amb els requisits de seguretat, toxicitat i compatibilitat de materials. El HFO-1234yf és molt menys perjudicial que el R-134a, però obliga a reconsiderar aspectes de disseny i seguretat. Alguns fabricants han expressat preocupació per la seva inflamabilitat en cas de col·lisió. La química dels refrigerants continua sent, per tant, un equilibri delicat entre rendiment, seguretat i sostenibilitat.
Una alternativa no inflamable és el refrigerant R-513A, una mescla azeotròpica de R-134a i R-1234yf, amb un GWP de 631 i classificació A1. Encara que el seu impacte ambiental és superior al de les HFO pures, ofereix una opció segura i eficient per substituir el R-134a en sistemes que no poden permetre riscos d’inflamació.
La Figura 4.22 mostra l’estructura de l’HFO-1234yf i la compara amb isòmers relacionats de l’HFO-1234ze.
4.5.5 Frenada i dissipació de calor
Frenar un vehicle significa eliminar la seva energia cinètica, i fer-ho amb rapidesa és essencial tant per seguretat com per rendiment. Si es disposés de prou temps, la fricció dels pneumàtics, del motor, de la transmissió i dels diferencials acabaria dissipant aquesta energia. Però una frenada intensa exigeix convertir-la gairebé instantàniament en una forma d’energia que es pugui evacuar de manera segura.
Els frens de disc moderns ho fan mitjançant la fricció, transformant l’energia cinètica de les rodes en calor ( ), que després es dissipa cap a l’entorn:
), que després es dissipa cap a l’entorn:
![\[ E_k \xrightarrow {\text {fricció}} Q \xrightarrow {\text {radiació}} \text {Atmosfera} \]](web_GEAForcesInt-images/image-255.svg)
Tecnologia de Frenada Regenerativa
En la frenada regenerativa, un motor elèctric utilitzat per impulsar les rodes es converteix en generador. La resistència de l’eix en gir converteix part de l’energia cinètica en energia elèctrica, que es pot emmagatzemar per ser utilitzada més endavant:
![\[ E_k \xrightarrow {\text {generació}} E_{\text {elèctrica}} \]](web_GEAForcesInt-images/image-256.svg)
Tot i que la frenada regenerativa millora l’eficiència, no és suficient per a una frenada d’emergència ràpida. Per a situacions crítiques, el sistema més efectiu és el de disc i pistó.
Mecanisme de frenada en vehicles amb disc
Cada roda equipada amb frens de disc porta un rotor metàl·lic solidari amb l’eix. Quan el conductor prem el pedal, la pressió hidràulica empeny les pastilles contra aquest rotor i apareix la fricció responsable de la frenada.
![\[ \text {Frenada} \Rightarrow \text {Pressió hidràulica} \Rightarrow \text {Pastilles de fre} \Rightarrow \text {Fricció} \Rightarrow \text {Calor (Q)} \]](web_GEAForcesInt-images/image-257.svg)
4.5.6 Material de fricció
Els materials de fricció utilitzats en vehicles comercials no són substàncies simples, sinó formulacions complexes. Hi trobem components durs, com l’alúmina, la sílice o altres ceràmiques, que aporten fricció; càrregues inerts, com argiles laminars; lubricants sòlids, com el grafit; i un aglutinant orgànic, sovint basat en resines fenòliques o en resorcinol.
Resines fenòliques
Les resines fenòliques polimeritzen a altes temperatures i originen una xarxa que proporciona cohesió mecànica i resistència tèrmica al material de la pastilla. Simplificant molt, el procés es pot descriure amb les reaccions següents:
![\[ \ch {C6H5OH} + \ch {CH2O} \rightarrow \ch {HOC6H4CH2OH} \]](web_GEAForcesInt-images/image-258.svg)
![\[ \ch {HOC6H4CH2OH} + \ch {C6H5OH} \rightarrow \ch {(HOC6H4)2CH2} + \ch {H2O} \]](web_GEAForcesInt-images/image-259.svg)
![\[ 2 \ch {HOC6H4CH2OH} \rightarrow \ch {(HOC6H4CH2)2O} + \ch {H2O} \]](web_GEAForcesInt-images/image-260.svg)
La Figura 4.23 mostra rutes representatives de formació de resines fenòliques a partir de fenol i formaldehid.
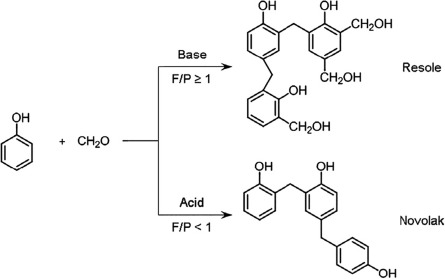
En aquestes etapes, el fenol ( ) reacciona amb el formaldehid (
) reacciona amb el formaldehid ( ) i es van formant ponts entre unitats orgàniques. Com més reticulada és l’estructura final, més gran és la resistència tèrmica i mecànica del material.
) i es van formant ponts entre unitats orgàniques. Com més reticulada és l’estructura final, més gran és la resistència tèrmica i mecànica del material.
L’aglutinant pot dissipar energia per diversos camins. Un dels més plausibles és la vaporització parcial o la reorganització de fragments del material, processos que exigeixen superar interaccions intermoleculars com ponts d’hidrogen i forces dispersives. En polímers de fenol/resorcinol, aquestes interaccions es mouen en un interval d’energia de 4 kJ/mol a 40 kJ/mol, compatible amb l’escala de calor generada en la fricció.
Un altre mecanisme possible per a l’energia tèrmica és la ruptura dels enllaços químics del lligant. Els polímers de fenol () i resorcinol ( ) contenen diversos tipus d’enllaços, com ara
) contenen diversos tipus d’enllaços, com ara  ,
,  ,
,  ,
,  ,
,  i
i  . Les energies mitjanes de dissociació d’aquests enllaços són les següents:
. Les energies mitjanes de dissociació d’aquests enllaços són les següents:
-
•
: 317 kJ/mol a 376 kJ/mol
-
•
: 728,3 kJ/mol
-
•
:  460 kJ/mol
460 kJ/mol
-
•
: 208 kJ/mol a 264 kJ/mol
-
•
: 361,9 kJ/mol
-
•
: 473,1 kJ/mol
Per a la ruptura d’aquests enllaços es necessitaria una gran quantitat de calor. Tanmateix, és possible que es generin temperatures localment elevades prop dels inductors de fregament en el material, convertint aquesta ruptura d’enllaços en un mecanisme menor de dissipació de calor.
També és possible que la calefacció del material provoqui reaccions entre l’aglutinant orgànic i l’oxigen de l’atmosfera. Aquest procés, essencialment una combustió, requereix aconseguir temperatures prou altes per superar l’energia d’activació necessària per a aquesta reacció. Tot i que la combustió de les pastilles de fre normalment no representa un problema, pot esdevenir-ho durant frens intensos a llarg termini, com en llargues baixades de vehicles pesants en terreny muntanyós.
Materials de fregament
La química també és decisiva en els materials utilitzats als rotors. Igual que passa amb els materials del radiador, el rotor ha de poder absorbir calor ràpidament i cedir-la després a l’atmosfera sense trencar-se, deformar-se ni perdre prestacions. Per millorar-ne la refrigeració i afavorir l’evacuació dels gasos generats al contacte amb la pastilla, alguns rotors incorporen ranures o perforacions.
Tradicionalment, els rotors, tant sòlids com perforats o ranurats, es fabriquen d’acer. En aplicacions d’alt rendiment, però, també s’utilitzen ceràmiques o fibres de carboni per reduir pes i mantenir un bon comportament a temperatures molt elevades. Aquest avantatge té un cost: quan estan freds, aquests materials acostumen a frenar pitjor que l’acer convencional. Per això s’empren sobretot en vehicles de prestacions altes i en competició.
El disseny i la química dels materials avançats dels rotors són temes àmpliament tractats en textos i articles de recerca sobre enginyeria de materials. Els materials de fricció i els rotors són exemples de com la química i la física es combinen per crear solucions eficients i efectives per a les necessitats del món real. La comprensió d’aquests materials és fonamental per al disseny de vehicles segurs i eficients, així com per a la millora contínua de les tecnologies de frenada.
4.6 Exercicis
4.6.1 Compendi d’exercicis
Exercici 12. Equació de Born-Mayer
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Dedueix l’equació de Born-Mayer a partir de considerar, de forma simplificada, que l’energia d’atracció Coulòmbica es pot expressar com i que la repulsió entre ions es pot expressar com .
Exercici 13. Energia de malla
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
L’òxid de magnesi, , té la mateixa estructura que el . Sabent que i que , calcula l’energia de malla d’aquest compost iònic.
Exercici 14. Identificació d’energies en processos de Born-Haber
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Anomena la magnitud energètica  corresponent a cadascun dels processos següents:
corresponent a cadascun dels processos següents:
-
1.

-
2.

-
3.

Exercici 15. Cicle de Born-Haber de l’òxid de calci
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Considera la formació de  a partir dels elements en el seu estat estàndard:
a partir dels elements en el seu estat estàndard:
![\[ \ch {Ca(s)} + \tfrac {1}{2}\ch {O2(g)} -> \ch {CaO(s)} \]](web_GEAForcesInt-images/image-289.svg)
Es disposa de les dades següents:
| Procés | (kJ/mol) |
| Entalpia estàndard de formació de |
−636 kJ/mol |
Sublimació de  |
192 kJ/mol |
Primera energia d’ionització de  |
590 kJ/mol |
| Segona energia d’ionització de |
1 145 kJ/mol |
Dissociació de  |
494 kJ/mol |
Primera afinitat electrònica de  |
−141 kJ/mol |
| Segona afinitat electrònica de |
845 kJ/mol |
-
1. Escriu els passos del cicle de Born-Haber per a la formació del
.
-
2. Calcula l’entalpia de formació dels ions gasosos
 i
i  a partir dels elements.
a partir dels elements.
-
3. Calcula l’entalpia de malla del
 .
.
Exercici 16. Cicle de Born-Haber de l’òxid de cesi
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Considera la formació de  a partir dels elements en el seu estat estàndard:
a partir dels elements en el seu estat estàndard:
![\[ \ch {2 Cs(s)} + \tfrac {1}{2}\ch {O2(g)} -> \ch {Cs2O(s)} \]](web_GEAForcesInt-images/image-303.svg)
Es disposa de les dades següents:
| Procés | (kJ/mol) |
| Entalpia estàndard de formació de |
−233 kJ/mol |
Sublimació de  |
78 kJ/mol |
Primera energia d’ionització de  |
375 kJ/mol |
| Dissociació de |
494 kJ/mol |
| Primera afinitat electrònica de |
−141 kJ/mol |
| Segona afinitat electrònica de |
845 kJ/mol |
-
1. Escriu els passos del cicle de Born-Haber per a la formació del
.
-
2. Calcula l’entalpia de formació dels ions gasosos
 i a partir dels elements.
i a partir dels elements.
-
3. Calcula l’entalpia de malla del
 .
.
Exercici 17. Cicle de Born-Haber del clorur d’estronci (EX4a, 15/04/2026)
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Considera la formació de  a partir dels elements en el seu estat estàndard:
a partir dels elements en el seu estat estàndard:
![\[ \ch {Sr(s)} + \ch {Cl2(g)} -> \ch {SrCl2(s)} \]](web_GEAForcesInt-images/image-316.svg)
-
1. Escriu els passos del cicle de Born-Haber per a la formació del
.
-
2. Calcula l’entalpia de formació dels ions gasosos
 i
i  a partir dels elements.
a partir dels elements.
-
3. Calcula l’entalpia estàndard de formació del
.
Es disposa de les dades següents:
| Procés | (kJ/mol) |
Sublimació de  |
164 kJ/mol |
Primera energia d’ionització de  |
549 kJ/mol |
| Segona energia d’ionització de |
1 064 kJ/mol |
Dissociació de  |
243 kJ/mol |
Afinitat electrònica de  |
−349 kJ/mol |
| Entalpia de malla de |
−2 150 kJ/mol |
Exercici 18. Intersticis en el coure FCC i defectes de Frenkel
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
El coure metàl·lic cristal·litza en una estructura cúbica centrada a les cares (FCC) amb paràmetre de cel·la  .
.
-
1. Calcula el radi metàl·lic del Cu suposant que els àtoms es toquen al llarg de la diagonal d’una cara.
-
2. Calcula el radi màxim d’un àtom que podria ocupar, sense distorsionar la xarxa ideal, un interstici octaèdric i un interstici tetraèdric.
-
3. Decideix si el Zn podria ocupar aquests intersticis. Proposa un àtom més raonable i indica si el cas correspon pròpiament a un defecte de Frenkel.
Exercici 19.  gasos ideals
gasos ideals
Perquè en una taula de pressió de vapor típicament no apareix la de l’,  i ?
i ?
Exercici 20. Sobreescalfar un líquid
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
És possible que un líquid arribi a estar sobreescalfat: temperatura superior a la d’ecullició per a aquella pressió però encara estat líquid, la qual cosa succeeix quan és molt pur i no hi ha partícules de pols. Com aconseguiries que no es produeixi aquest sobreecalfament?
Exercici 21. Aigua gas-líquida
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Què ens produirà una cremada més gran: una massa d’(g) a 100 graus o la mateixa quantitat d’aigua líquida a la mateixa temperatura?
Exercici 22. Simulació del moviment brownià
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Usant R, prova d’executar aquest script que mostra com simular el moviment Brownià d’una partícula en un líquid (extret de http://www.phytools.org/eqg/phytools/):
1 t <− 0:100 # temps de simulacio 2 sig2 <− 0.01 3 ## primer , calcula un conjunt de desviacions aleatòries puntuals 4 x <− rnorm ( n = length ( t ) − 1, sd = sqrt ( sig2 ) ) 5 ## després , acumula'n els resultats 6 x <− c (0, cumsum(x) ) 7 plot ( t , x , type = ” l ” , ylim = c (−2, 2) )
Després, executa el següent script, que produeix 10000 simulacions diferents:
1 nsim <− 1000 2 ## creo una matriu que hostatgi totes les simulacions 3 X <− matrix (0, nsim , length ( t ) ) 4 for ( i in 1: nsim ) X[ i , ] <− c (0, cumsum(rnorm(n = length ( t ) − 1, sd = sqrt ( sig2 ) ) ) ) 5 plot ( t , X [1, ], xlab = ” temps ” , ylab = ” desviacions ” , ylim = c (−2, 2) , type = ” l ” ) 6 for ( i in 1: nsim ) lines ( t , X[ i , ])
Exercici 23. Variància de simulacions brownianes
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Per saber la variança que s’obté de la simulació podem fer
1 var ( X [, length ( t ) ])
i per mostrar l’histograma de posicions finals:
1 hist ( X [, length ( t ) ])
o bé:
1 plot ( density ( X [, length ( t ) ]) )
Calcula la variança de la distribució per a diferents valors del nombre de simulacions o el temps simulat.
Qüestió 24. Perquè es generen bombolles de seguida que obrim una ampolla d’aigua amb gas?
Qüestió 25. La constant de la llei de Henry de l’ en aigua a 25 °C és 1,27 · 10−3 mol/(L atm), i la fracció molar de l’ en l’atmosfera és 0,21. Calcula la solubilitat de l’ en aigua a 25 °C i a pressió atmosfèrica.
Exercici 26. Ebullició aigua
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
En un recipient hi ha aigua líquida. Es connecta el recipient a una bomba de buit i es va abaixant la pressió sobre el líquid. Si la temperatura és de 60 graus, a quina pressió bullirà l’aigua?
Qüestió 27. La de congelació del benzè pur és 5.40°C. Quan es dissol 1.15g de naftalè en 100 g de benzè, la dissolució resultant té un punt de congelació de 4.95°C. Si la constant de descens molal del punt de congelació del benzè és 5.12°C, quin és el pes molecular del naftalè?
Exercici 28. No idealitat i punt d’ebullició
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Raona l’efecte que té la no idealitat de les dissolucions segons la Figura ??a a) en el seu punt d’ebullició, i b) en un procés de destil·lació fraccionada.
Exercici 29. Azeòtrops i destil·lació
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Un azeòtrop positiu prové d’una desviació també positiva de la llei de Raoult. a) Dibuixa la corba de Temperatura d’ebullició vs composició per a un azeòtrop positiu basant-te en les Figures ??a i ??a. b) Raona el resultat de fer una destil·lació a partir de diverses composicions d’aquesta mescla. c) què succeiria en un azeòtrop negatiu?
Exercici 30. Destil·lació pressure-swing
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Volem separar una barreja equimolar d’etanol i acetat etílic per destil·lació en productes relativament purs. La barreja forma un azeòtrop de mínim punt d’ebullició segons la Figura ??. No obstant, la composició de l’azeòtrop és sensible a la pressió, mostrant un increment significatiu de la fracció molar de l’etanol quan incrementa la pressió, com es mostra a la Figura. Dibuixa un esquema aproximat per a la separació de les dues components de la barreja que tregui profit d’aquest fet.
Exercici 31. PM naftalè
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
La temperatura de congelació del benzè pur és 5,40 °C i la seva constant crioscòpica és 5,12 °C/(mol kg). Si es dissolen 1,15 g de naftalè en 100 g de benzè, la dissolució resultant congela a 4,95 °C. Calcula el pes molecular del naftalè.
Exercici 32. Descens de la pressió de vapor en una dissolució ideal
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Una quantitat  d’un solut no volàtil es dissol en
d’un solut no volàtil es dissol en  mols d’un dissolvent volàtil. Suposa que la dissolució és ideal i que, a la temperatura considerada, la pressió de vapor del dissolvent pur és
mols d’un dissolvent volàtil. Suposa que la dissolució és ideal i que, a la temperatura considerada, la pressió de vapor del dissolvent pur és  .
.
-
1. Escriu la pressió de vapor de la dissolució segons la llei de Raoult.
-
2. Dedueix l’expressió del descens absolut de pressió de vapor,
 , en funció de la fracció molar del solut.
, en funció de la fracció molar del solut.
-
3. Dedueix també el descens relatiu de pressió de vapor,
 .
.
Exercici 33. Mescla etanol-metanol[12]
L’etanol i el metanol formen una dissolució aproximadament ideal. La pressió de vapor de l’etanol és de 44,5 mmHg i la del metanol és de 88,7 mmHg a 20 °C.
-
a) Trobar les fraccions molars del metanol i l’etanol en una dissolució que s’obté mesclant 60 g d’etanol amb 40 g de metanol.
-
b) Trobar les pressions parcials i la pressió de vapor total de la dissolució, així com la fracció molar de l’etanol en el vapor.
Exercici 34. Dissolució benzè-toulè[12]
A 20 °C, la pressió de vapor del benzè pur és 75 mmHg i la del toluè pur de 22 mmHg. Quina és la composició d’una dissolució d’aquests dos components, que té una pressió de vapor de 50 mmHg a aquesta temperatura? Quina serà la composició del vapor en equilibri d’aquesta dissolució?
Exercici 35. Determinació de la massa molecular del fòsfor vermell (Exercici 4 2025)
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Es dissolen 0,30 g de fòsfor en 500 g d’etanol. La dissolució obtinguda presenta un punt d’ebullició 0,005 9 °C superior al de l’etanol pur.
Determina la massa molar del fòsfor dissolt i dedueix quina fórmula molecular li correspon.
(Dades: constant ebullioscòpica de l’etanol  , massa atòmica del fòsfor
, massa atòmica del fòsfor  )
)
Exercici 36. Solubilitat del bòrax[12]
La solubilitat del bòrax ( ) a l’aigua augmenta a mesura que augmenta la temperatura. Quan la sal es dissol, es desprèn o s’absorbeix calor? És l’entalpia del procés de dissolució positiva o negativa?
) a l’aigua augmenta a mesura que augmenta la temperatura. Quan la sal es dissol, es desprèn o s’absorbeix calor? És l’entalpia del procés de dissolució positiva o negativa?
Exercici 37. Desviació de la llei de Raoult[12]
A 55 °C, l’etanol té una pressió de vapor de 168 mmHg, i la pressió de vapor del metil ciclohexà és de 280 mmHg. Una dissolució dels dos components, en la qual la fracció molar de l’etanol és  , té una pressió de vapor total de 476 mmHg. És el procés de formació de la dissolució exotèrmic o endotèrmic?
, té una pressió de vapor total de 476 mmHg. És el procés de formació de la dissolució exotèrmic o endotèrmic?
Exercici 38. Azeòtrops
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Un azeòtrop positiu prové d’una desviació també positiva de la llei de Raoult.
-
1. Dibuixa la corba de Temperatura d’ebullició vs composició per a un azeòtrop positiu basant-te en les Figures ??a i ??a.
-
2. Raona el resultat de fer una destil·lació a partir de diverses composicions d’aquesta mescla.
-
3. Què succeiria en un azeòtrop negatiu?
Exercici 39. Vaporització de l’aigua en un recipient tancat
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
La pressió de vapor de l’aigua a 25 °C és de 23,76 torr. Si es tanquen 1,25 g d’aigua en un recipient de 1,5 L, hi haurà algun líquid present? Si és així, quina massa de líquid?
Exercici 40. Diagrama de fases de l’aigua
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Segons la imatge:
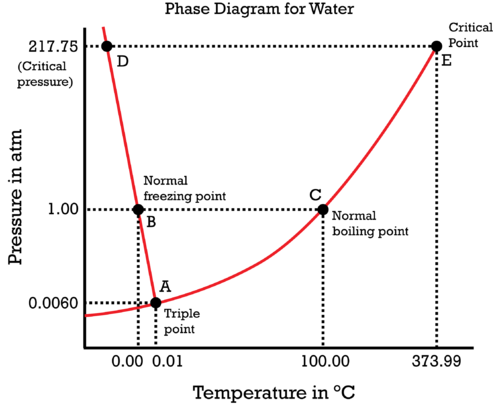
-
1. Quina és la temperatura de fusió de l’aigua a 1 atm?
-
2. Quina és la temperatura d’ebullició de l’aigua a 1 atm?
-
3. Quina és la pressió de vapor de l’aigua a 25 °C?
-
4. Quina és la pressió crítica de l’aigua?
-
5. Quina és la temperatura crítica de l’aigua?
-
6. A quines condicions de temperatura i pressió es troba l’aigua en estat líquid, sòlid i gasós, tots tres en equilibri?
-
7. Determina si l’aigua és sòlida, líquida o gasosa en cadascuna de les següents combinacions de temperatura i pressió:
-
• 1 atm i 20 °C
-
• 0.006 atm i 100 °C
-
• 210 atm i 100 °C
-
• 10 atm i 50 °C
-
Adaptat de Libretexts Chemistry.
Exercici 41. Termodinàmica de la vaporització de l’aigua
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Calcular els canvis d’energia lliure, entalpia i entropia per a la vaporització de l’aigua líquida a la temperatura de 100 °C i 1 atm de pressió, sabent que la calor de vaporització és 40,7 kJ/mol.
Exercici 42. Manteniment d’un material a temperatura controlada
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Un material molt sensible ha de mantenir-se a una temperatura propera a 0 °C per evitar-ne la degradació. Es disposa únicament d’aigua i de metanol (tots dos a temperatura ambient) com a líquids, i de nitrogen líquid i gel sec (diòxid de carboni sòlid) com a agents refrigerants.
Quina combinació de líquid i agent refrigerant s’hauria d’utilitzar per crear un bany de refrigeració adequat? Justifica la resposta, sabent que:
-
•
 sublima a −78,5 °C.
sublima a −78,5 °C.
-
•
 bull a −195,8 °C.
bull a −195,8 °C.
-
• L’aigua congela a 0 °C.
-
• El metanol congela a −143,7 °C.
Exercici 43. Equilibri de síntesi i hidròlisi del ZDDP
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
El zinc dialquilditiofosfat (ZDDP) és una família d’additius antidesgast emprats en olis lubricants. De manera simplificada, es pot obtenir en dues etapes:
-
1. Formació de l’àcid dialquilditiofosfòric, representat aquí amb grups metil:
-
2. Neutralització de l’àcid amb òxid de zinc per formar el zinc bis(dialquilditiofosfat):

![(4.6.2) \begin{equation} \ch {2 (CH3O)2P(S)SH + ZnO(s) <=> Zn[(CH3O)2P(S)S]2 + H2O} \label {eq:zddp_synthesis} \end{equation}](web_GEAForcesInt-images/image-352.svg)
Fixem-nos en la Reacció 4.6.2. Per simplificar, anomenem
![\[ \mathrm {HA}=\ch {(CH3O)2P(S)SH} \qquad \mathrm {ZnA_2}=\ch {Zn[(CH3O)2P(S)S]2} \]](web_GEAForcesInt-images/image-353.svg)
-
1. Escriu l’expressió de la constant d’equilibri en termes d’activitats.
-
2. Escriu l’expressió aproximada de
 si
si  ,
,  i
i  es troben dissolts en la mateixa fase orgànica.
es troben dissolts en la mateixa fase orgànica.
-
3. Explica qualitativament com afecta la presència d’aigua al desplaçament de l’equilibri i a la quantitat de ZDDP disponible.
-
4. Relaciona aquest efecte amb la capacitat del lubricant de protegir les superfícies metàl·liques.
Exercici 44. Calor de dissolució del 
En un experiment de calorimetria, es vol determinar la calor de dissolució del .
-
• Es va fer passar un corrent de 1,03 A durant 23 s a través d’una resistència amb una diferència de potencial de 12,00 V.
-
• Durant aquest procés, la temperatura de l’aigua del calorímetre va augmentar de 20,340 °C a 22,340 °C.
-
• Posteriorment, es van dissoldre 2,45 g de
sòlid en el calorímetre, provocant una pujada de temperatura de 20,34 °C a 26,22 °C.
-
1. Calcula el factor de calibratge del calorímetre en J/°C (el factor de calibratge és la quantitat d’energia (en joules) necessària per augmentar la temperatura del calorímetre en 1°C).
-
2. Calcula la variació d’entalpia (
 ) de dissolució del en kJ/mol.
) de dissolució del en kJ/mol.
Exercici 45. Entalpia de dissolució del clorur de calci
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Quan el clorur de calci anhidre () es dissol en aigua, es produeixen diversos canvis d’energia. Considera les dades següents:
-
• Entalpia de xarxa de
: 2 258 kJ/mol
-
• Entalpia d’hidratació de
 : −1 650 kJ/mol
: −1 650 kJ/mol
-
• Entalpia d’hidratació de
 : −364 kJ/mol
: −364 kJ/mol
-
1. Escriu el cicle de Hess que descriu aquest procés.
-
2. Calcula l’entalpia de dissolució (
 ) de .
) de .
-
3. Reflexiona sobre les diferències entre el resultat d’aquest exercici i el de l’Exercici 44.
Exercici 46. Diagrama de fases amb bucle tancat [5]
La figura mostra un diagrama de fases de bucle tancat per a una mescla líquida binària  , en funció de la temperatura i de la fracció molar
, en funció de la temperatura i de la fracció molar  .
.
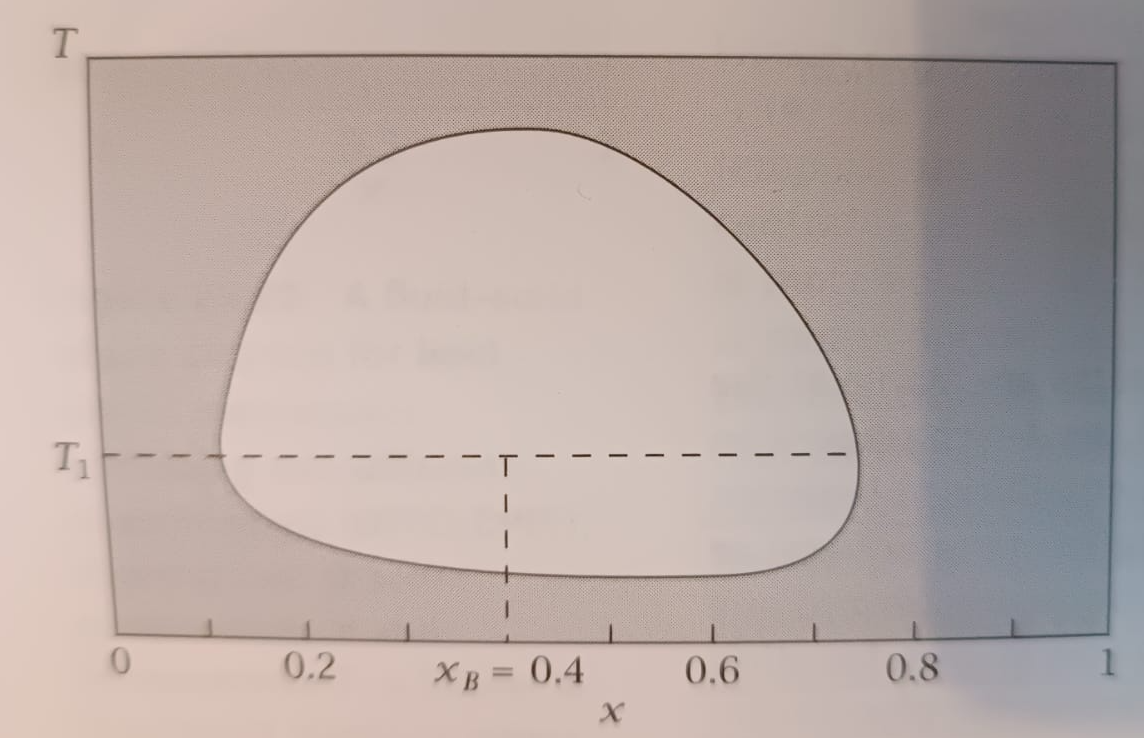
Per a una mescla amb composició global  a la temperatura
a la temperatura  :
:
-
1. Estima les composicions
 i
i  de les dues fases en equilibri.
de les dues fases en equilibri.
-
2. Si hi ha
 de solució, estima els volums de les fases rica en i rica en
de solució, estima els volums de les fases rica en i rica en  .
.
Exercici 47. Diagrama de fases de l’àcid úric [5]
La figura mostra un diagrama de fases simplificat de l’àcid úric en sang. La zona grisa és una fase i la zona blanca correspon a coexistència de dues fases.
-
1. Estima el límit de solubilitat de l’àcid úric a
 i a
i a  .
.
-
2. Explica per què el refredament pot afavorir atacs de gota.
Exercici 48. Funcions d’energia lliure i solidificació [5]
La figura mostra un diagrama de fases general d’un procés de solidificació en una mescla binària.

Descriu qualitativament quina forma han de tenir les corbes d’energia lliure  de les fases líquida i sòlida per donar lloc a les regions d’una fase i de coexistència líquid/sòlid.
de les fases líquida i sòlida per donar lloc a les regions d’una fase i de coexistència líquid/sòlid.
Exercici 49. Mescla segons el model reticular [5]
Una solució conté molècules i a  . Suposa que cada dos contactes
. Suposa que cada dos contactes  són menys estables que un contacte
són menys estables que un contacte  més un contacte
més un contacte  en una energia
en una energia  , i que cada molècula té de mitjana sis veïns. Per a
, i que cada molècula té de mitjana sis veïns. Per a  , a quina temperatura cal escalfar el sistema perquè es dissolgui completament en ?
, a quina temperatura cal escalfar el sistema perquè es dissolgui completament en ?
Exercici 50. Binodal i espinodal en el model reticular [5]
Considera una mescla reticular simètrica a una temperatura per a la qual  .
.
-
1. Calcula les composicions binodals
i .
-
2. Calcula les composicions espinodals.
-
3. Indica els intervals de composició en què la solució és metastable.
Exercici 51. Bullida d’una mescla acetona-ciclohexà [5]
La figura mostra l’equilibri líquid-vapor de mescles d’acetona en ciclohexà.
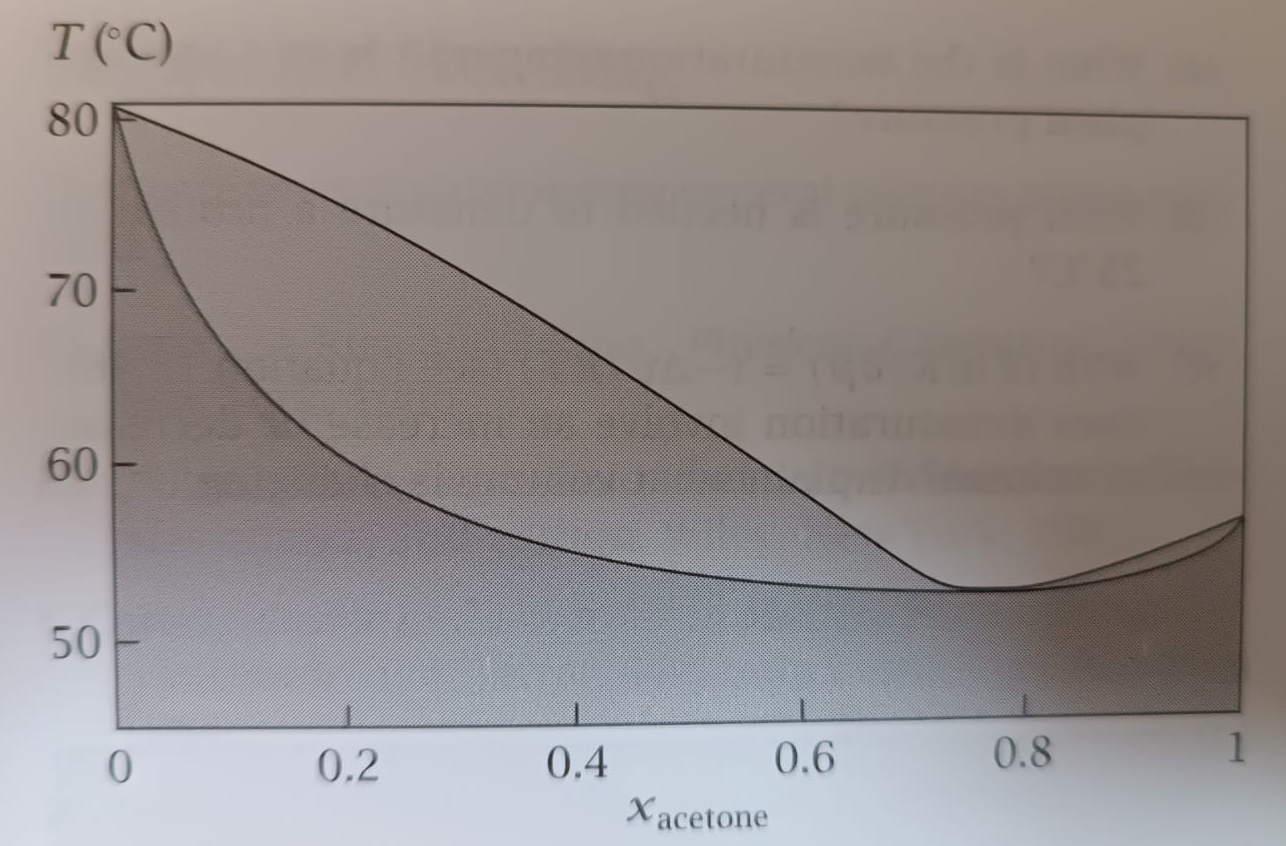
Descriu qualitativament què és diferent en la bullida d’un líquid amb  respecte d’un líquid amb
respecte d’un líquid amb  .
.
Exercici 52. Azeòtrop etanol-aigua [5]
La figura mostra un diagrama de fases de bullida per a mescles alcohol-aigua. L’azeòtrop és el punt on  i
i  .
.
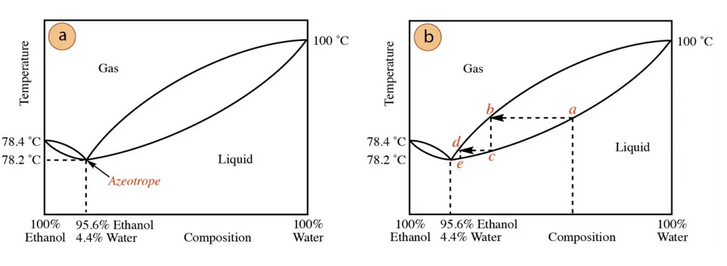
Descriu què representa el punt azeotròpic i què implica per a la destil·lació d’alcohol.
Exercici 53. Solubilitats per determinar  [5]
[5]
La solubilitat de l’etilbenzè en aigua és  a
a  , i la del propilbenzè en aigua a la mateixa temperatura és
, i la del propilbenzè en aigua a la mateixa temperatura és  .
.
-
1. Calcula el paràmetre d’interacció
 per a l’etilbenzè en aigua.
per a l’etilbenzè en aigua.
-
2. Calcula el paràmetre
per al propilbenzè en aigua.
-
3. Estima l’energia lliure de transferir un grup
 d’un hidrocarbur a l’aigua a .
d’un hidrocarbur a l’aigua a .
Exercici 54. Temperatures crítiques inferior i superior de solubilitat [5]
La figura mostra tres diagrames de fases líquid-líquid: aigua-trietilamina, aigua-2-metilpiperidina i sofre-benzè.
Explica què indiquen aquests diagrames sobre les interaccions intermoleculars en cada sistema.
Exercici 55. Punts crítics de l’aigua i del diòxid de carboni [5]
Les constants de van der Waals són  i
i  per al
per al  , i
, i  i
i  per a l’. Calcula i
per a l’. Calcula i  per a tots dos fluids i explica per què el és més pràctic en cromatografia de fluids supercrítics.
per a tots dos fluids i explica per què el és més pràctic en cromatografia de fluids supercrítics.
4.6.2 Exercicis d’autoavaluació
Exercici 56. Ratio d’empaquetament
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
La ratio d’empaquetament d’una cel·la unitat es defineix com la fracció entre el volum omplert pels àtoms que la formen i el seu volum total. Calcula el RE de la cel·la unitat cúbica centrada en la cara i de la cel·la unitat cúbica centrada en el cos.
Exercici 57. Interacció Coulòmbica
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
L’energia potencial Coulòmbica entre dos ions es pot expressar com:

on  és la càrrega de l’ió,
és la càrrega de l’ió,  és la permitivitat del buit i és la distància entre els ions.
és la permitivitat del buit i és la distància entre els ions.
Se sap que una molècula gasosa de té una distància interatòmica de 2.38Å. Quina és l’energia potencial Coulòmbica d’un mol d’aquestes molècules?
Exercici 58. Born-Haber
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Calcula l’energia reticular del Fluorur de Liti usant l’esquema del cicle de Born-Haber que en trobaras a la Wikipedia:
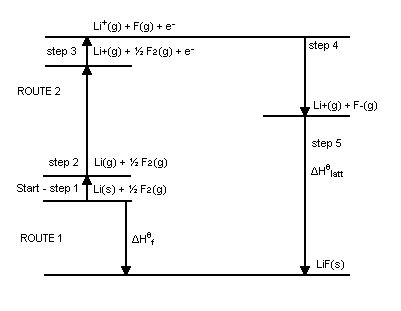
sabent que:
-
• L’energia de sublimació del liti és 161 kJ/mol.
-
• L’energia d’ionització del liti és 520 kJ/mol.
-
• L’energia de dissociació del fluor és 158 kJ/mol.
-
• L’afinidat electrònica del fluor és −328 kJ/mol.
-
• L’energia de formació del compost és −617 kJ/mol.
Exercici 59. Propietats sòlids
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Compara, per a diferents tipus de sòlids (covalents, iònics, metàl·lics o moleculars), les següents característiques: pressió de vapor, punt de fusió, punt d’ebullició, duresa, fragilitat, conducció elèctrica en estat sòlid, i conducció elèctrica en estat líquid.
Exercici 60. Distància d’enllaç C-C
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
L’estructura cristal·lina del diamant és equivalent a una xarxa cúbica centrada en la cara (FCC), amb una base de dos àtoms de carboni idèntics: un a la posició  i l’altre a
i l’altre a  , on les coordenades es donen com a fraccions al llarg dels costats del cub. Això és equivalent a dues xarxes FCC interpenetrants, desplaçades una respecte a l’altra al llarg d’una diagonal del cos en un quart de la seva longitud. A la figura, els carbonis en els vèrtexs es mostren en color negre, els que
són a les cares en color vermell i els que són interiors en color blau.
, on les coordenades es donen com a fraccions al llarg dels costats del cub. Això és equivalent a dues xarxes FCC interpenetrants, desplaçades una respecte a l’altra al llarg d’una diagonal del cos en un quart de la seva longitud. A la figura, els carbonis en els vèrtexs es mostren en color negre, els que
són a les cares en color vermell i els que són interiors en color blau.
Si la densitat del diamant és 3,51 g/cm3 a 25 °C i 1,00 atm de pressió, calculeu la distància de l’enllaç C-C en aquest cristall covalent.
Exercici 61. Defectes de Frenkel i Schottky
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Si la densitat del clorur sòdic sense defectes és de 2,165 g/cm3, quina seria la densitat si tingués una fracció de defectes de  en els casos següents? Suposa que el volum no varia amb els defectes.
en els casos següents? Suposa que el volum no varia amb els defectes.
-
1. Defectes de Frenkel.
-
2. Defectes de Schottky, entenent
com el nombre de parelles de vacants  per cada parell iònic
per cada parell iònic  inicial.
inicial.
Exercici 62. Ebullició dissolucions
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Respon les següents preguntes:
-
1. Exactament 1.00g d’urea dissolts en 75.00g d’aigua donen una dissolució que bull a 100.114
 C. El pes molecular de la urea és 60.1. Quina és la de l’aigua?
C. El pes molecular de la urea és 60.1. Quina és la de l’aigua?
-
2. Una dissolució preparada dissolent 12,00g de glucosa en 100g d’aigua bull a 100.34
C. Quin és el pes molecular de la glucosa?
Exercici 63. Llei de Raoult
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Raona l’efecte d’un solut en la pressió de vapor d’un solvent, usant una visió macroscòpica o bé microscòpica.
Exercici 64. PM sucrosa
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
La pressió de vapor de l’aigua a 20 °C és 17,54 mmHg. Quan dissolem 114 g de sucrosa en 1 000 g d’aigua, la pressió de vapor es redueix en 0,11 mmHg. Quin és el pes molecular de la sucrosa?
Exercici 65. Solubilitat 
Quina és la constant de solubilitat del cromat d’argent ( ) si la concentració d’una dissolució saturada d’aquesta sal té una concentració de 6.7
) si la concentració d’una dissolució saturada d’aquesta sal té una concentració de 6.7 10
10 M d’ions cromat?
M d’ions cromat?
4.6.3 Exercicis d’examens anteriors
Exercici 66. Cicle de Born-Haber del clorur de potassi (Parcial 2, 2024-2025)
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
A partir de les dades de la taula,
| Procés | (kJ/mol) |
|
Sublimació de  |
+89 | |
| Dissociació de |
+243 | |
Potencial d’ionització de  |
+419 | |
| Afinitat electrònica de |
−349 | |
Descomposició  en els seus elements en l’estat de referència en els seus elements en l’estat de referència |
+437 |
-
1. calcula l’energia de formació de l’ió
 ;(0,5 punts)
;(0,5 punts)
-
2. calcula l’energia de formació de l’ió
;(0,5 punts)
-
3. calcula l’energia de formació del
;(0,5 punts)
-
4. calcula l’entalpia de malla del
;(1 punt)
Exercici 67. Raoult electròlits[12] (Parcial 2, 2024-2025)
Quan es dissolen 3,24 g de nitrat de mercuri,  , en 1 000 g d’aigua, la temperatura de congelació de la dissolució és −0,055 8 °C. Quan es dissolen 10,84 g de clorur de mercuri,
, en 1 000 g d’aigua, la temperatura de congelació de la dissolució és −0,055 8 °C. Quan es dissolen 10,84 g de clorur de mercuri,  , en 1 000 g d’aigua, la temperatura de congelació de la dissolució és −0,074 4 °C. La constant de congelació de l’aigua és 1,86 °C kg/mol.
, en 1 000 g d’aigua, la temperatura de congelació de la dissolució és −0,074 4 °C. La constant de congelació de l’aigua és 1,86 °C kg/mol.
-
1. Quina és la molalitat de cadascuna de les dues dissolucions?(1 punt)
-
2. Quina és la raó entre el número de mols de solut i el número de mols de solut per kg de dissolvent en els dos casos?(1 punt)
-
3. S’ha dissociat alguna de les dues sals?(0.5 punts)
Exercici 68. Calorimetria i dissolució de  (Parcial 3, 2024-2025)
(Parcial 3, 2024-2025)
En un experiment de calorimetria, es vol determinar la calor de dissolució del  . Es va fer passar un corrent de 0,950 A durant 35 s a través d’una resistència amb una diferència de potencial de 10,00 V. Durant aquest procés, la temperatura de l’aigua del calorímetre va augmentar de 21,15 °C a 23,05 °C. Posteriorment, es
van dissoldre 3,20 g de sòlid en el calorímetre, provocant una baixada de temperatura de 22,94 °C a 19,88 °C.
. Es va fer passar un corrent de 0,950 A durant 35 s a través d’una resistència amb una diferència de potencial de 10,00 V. Durant aquest procés, la temperatura de l’aigua del calorímetre va augmentar de 21,15 °C a 23,05 °C. Posteriorment, es
van dissoldre 3,20 g de sòlid en el calorímetre, provocant una baixada de temperatura de 22,94 °C a 19,88 °C.
-
1. Calcula el factor de calibratge del calorímetre en J/°C.(1 punt)
-
2. Quina és la calor absorbida per la dissolució del
en el calorímetre en J?(1 punt)
-
3. Calcula la variació d’entalpia (
) de dissolució del en kJ/mol.(1 punt)
-
4. Seria el
un bon substitut del per a l’eliminació del gel a les carreteres? Justifica la teva resposta.(1 punt)
Exercici 69. Recuperacions: dissolucions i propietats col.ligatives
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Els exercicis següents provenen d’exàmens de recuperació.
-
1. La pressió de vapor del benzè pur a 20 °C és de 75 Torr, i la del metilbenzè pur és de 25 Torr a la mateixa temperatura. Calcula la pressió de vapor d’una barreja equimolar de benzè i metilbenzè a 20 °C.
-
2. Amb les mateixes pressions de vapor, quina és la composició del vapor en equilibri amb una dissolució ideal on la fracció molar de benzè en el líquid és
 ?
?
-
3. El clorur d’amoni, a 298 K, es dissol en aigua dissociant-se en els seus ions. Es tracta d’un procés espontani?
Dades a 298 K:
![\[ \ch {NH4Cl_{(s)} -> NH4+_{(aq)} + Cl-_{(aq)}} \]](web_GEAForcesInt-images/image-455.svg)
![\[ \begin {array}{c|cc} \text {substància} & \Delta H_f^0\ /\ \si {\kilo \cal \per \mol } & S^0\ /\ \si {\cal \per \mol \per \kelvin } \\\hline \ch {NH4Cl_{(s)}} & -75.38 & 22.6 \\ \ch {NH4+_{(aq)}} & -31.74
& 26.97 \\ \ch {Cl-_{(aq)}} & -40.02 & 13.2 \end {array} \]](web_GEAForcesInt-images/image-456.svg)
Exercici 70. Cicle de Born-Haber del fluorur de magnesi (Parcial 2, 2025-2026)
Mostra la resolució
Resolució disponible a Exercise.pdf i als materials d autoavaluacio daquest tema.
Considera la formació de  a partir dels elements en el seu estat estàndard:
a partir dels elements en el seu estat estàndard:
![\[ \ch {Mg(s) + F2(g) -> MgF2(s)} \]](web_GEAForcesInt-images/image-458.svg)
Exercici adaptat a partir d’exercicis de cicles de Born-Haber de Chemistry LibreTexts.
Es disposa de les dades tabulades següents [11, 10, 9, 8]:
| Procés | (kJ/mol) |
| Entalpia estàndard de formació de |
−1 124 kJ/mol |
Sublimació de  |
148 kJ/mol |
Primera energia d’ionització de  |
738 kJ/mol |
| Segona energia d’ionització de |
1 451 kJ/mol |
Dissociació de  |
159 kJ/mol |
Afinitat electrònica de  |
−328 kJ/mol |
-
1. Escriu els passos del cicle de Born-Haber per a la formació del
.(0,5 punts)
-
2. Calcula l’entalpia de formació dels ions gasosos
 i
i  a partir dels elements.(1 punt)
a partir dels elements.(1 punt)
-
3. Calcula l’entalpia de malla del
.(0,5 punts)
-
4. Indica si la formació de la xarxa cristal·lina és endotèrmica o exotèrmica.(0,5 punts)
Exercici 71. Solubilitat de  amb la llei de Henry (Parcial 2, 2025-2026)
amb la llei de Henry (Parcial 2, 2025-2026)
Una ampolla rígida de 0,750 L conté aigua carbonatada a 25 °C. Abans d’obrir-la, la pressió parcial de sobre el líquid és de 4,0 atm. A aquesta temperatura, la constant de Henry per al en aigua és: Exercici adaptat a partir d’exercicis de llei de Henry i solubilitat de gasos de Chemistry LibreTexts.
![\[ k_\text {H}(\ch {CO2})=\qty {3.4e-2}{\mol \per \liter \per \atm } \]](web_GEAForcesInt-images/image-473.svg)
tabulada a [7], on  . Un cop oberta l’ampolla i assolida la nova situació d’equilibri amb l’aire, prenem
. Un cop oberta l’ampolla i assolida la nova situació d’equilibri amb l’aire, prenem  .
.
-
1. Calcula la concentració de
dissolt abans d’obrir l’ampolla.(0,5 punts)
-
2. Calcula els mols de
dissolts inicialment en els 0,750 L.(0,5 punts)
-
3. Calcula els mols i la massa de
que deixen d’estar dissolts després d’obrir l’ampolla.(1 punt)
-
4. Compara qualitativament aquest resultat amb el que passaria si la pressió parcial inicial fos només de 1,0 atm.(0,5 punts)
Bibliografia
-
[1] Geoffrey M. Bowers i Ruth A. Bowers. Understanding Chemistry through Cars. en. CRC Press, nov. de 2014. isbn: 978-1-4665-7184-6. 10.1201/b17581.
-
[2] Aureli Caamaño Ros, Armand Servent Tarragona i Damià Obach Muntada. Química, COU. spa. Teide, 1991. isbn: 978-84-307-3338-5. uRl: https://dialnet.unirioja.es/servlet/libro?codigo=53646 (cons. 09-08-2023).
-
[3] Chemical bonding | Definition, Types, & Examples | Britannica. en. Març de 2025. uRl: https://www.britannica.com/science/chemical-bonding (cons. 24-03-2025).
-
[4] Ken A. Dill i Sarina Bromberg. Molecular driving forces: statistical thermodynamics in biology, chemistry, physics, and nanoscience. eng. 2nd ed. Boca Raton, Florida: CRC press, Taylor & Francis group, 2011. isbn: 978-0-8153-4430-8.
-
[5] Ken A. Dill i Sarina Bromberg. Molecular Driving Forces: Statistical Thermodynamics in Chemistry and Biology. New York: Garland Science, 2003. isbn: 0-8153-2051-5.
-
[6] Ira N. Levine. Physical chemistry. eng. 5. ed., internat. ed., [Nachdr.] Boston: McGraw-Hill, 2003. isbn: 978-0-07-253495-5 978-0-07-231808-1 978-0-07-112242-9 978-0-07-123213-5.
-
[7] Aqueous Solubility and Henry’s Law Constants of Organic Compounds. A: CRC Handbook of Chemistry and Physics. Internet Version 2005. Ed. de David R. Lide. 85a ed. Boca Raton, FL: CRC Press, 2005.
-
[8] Bond Dissociation Energies in Simple Molecules. A: CRC Handbook of Chemistry and Physics. Internet Version 2005. Ed. de David R. Lide. 85a ed. Boca Raton, FL: CRC Press, 2005.
-
[9] Electron Affinities. A: CRC Handbook of Chemistry and Physics. Internet Version 2005. Ed. de David R. Lide. 85a ed. Boca Raton, FL: CRC Press, 2005.
-
[10] Ionization Potentials of Atoms and Atomic Ions. A: CRC Handbook of Chemistry and Physics. Internet Version 2005. Ed. de David R. Lide. 85a ed. Boca Raton, FL: CRC Press, 2005.
-
[11] Standard Thermodynamic Properties of Chemical Substances. A: CRC Handbook of Chemistry and Physics. Internet Version 2005. Ed. de David R. Lide. 85a ed. Boca Raton, FL: CRC Press, 2005.
-
[12] Bruce H. Mahan. QUIMICA Curso Universitario. Español. 1977.
-
[13] S Soni, J Singh i B K Gill. “Role of nanofluids as an anti-freezing agent”. en. A: IOP Conf. Ser.: Mater. Sci. Eng. 1225.1 (febr. de 2022). Publisher: IOP Publishing, pàg. 012049. issn: 1757-899X. 10.1088/1757-899X/1225/1/012049.
-
[14] Paul Peter Urone i Roger Hinrichs. College Physics. Ed. d’OpenStax. 2012. uRl: http://cnx.org/contents/031da8d3-b525-429c-80cf-6c8ed997733a@11.1..
-
[15] Hao Wu i Joseph H. Koo. “Chapter 8 - Characterization of high-temperature polymers for extreme environments”. A: Analysis of Flame Retardancy in Polymer Science. Ed. d’Henri Vahabi, Mohammad Reza Saeb i Giulio Malucelli. Elsevier, gen. de 2022, pàg. 299 - 331. isbn: 978-0-12-824045-8. 10.1016/B978-0-12-824045-8.00008-3.
-
[16] Teh Fu Yen. Chemistry For Engineers. en. Google-Books-ID: FM42DwAAQBAJ. World Scientific, gen. de 2008. isbn: 978-1-911298-41-0.